


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
VALIDATION ESTIMATION OF THE CHROMATOGRAPHIC METHOD FOR DETERMINATION OF ANILOCAIN IN BLOOD PLASMA

Изучение фармакокинетических параметров потенциального препарата, одного из важнейших этапов доклинических и клинических исследований, невозможно без разработки высокочувствительных, точных и воспроизводимых методик определения его в биологических объектах. Лидирующее место среди методов определения лекарственных средств в биологическом материале, в том числе при изучении фармакокинетики, занимает высокоэффективная жидкостная хроматография (ВЭЖХ) с различными видами детектирования [1, 3]. Микроколоночная ВЭЖХ в силу дополнительных преимуществ, таких как экспрессность, экономичность, удовлетворительные метрологические характеристики в настоящее время активно используется для решения сложных медицинских задач в области лекарственного терапевтического мониторинга и фармакокинетических исследований [6]. В связи с этим, целью настоящей работы явилась разработка и валидация методики определения анилокаина в плазме крови методом микроколоночной обращённо-фазной ВЭЖХ для целей изучения фармакокинетики.
Материалы и методы исследования
В работе использовали микроколоночный жидкостный хроматограф "Милихром А- 02" (ЗАО ИХ "ЭкоНова", Новосибирск, Россия) с колонкой размером 2х75 мм, заполненной обращено-фазным сорбентом ProntoSIL 120-5C18 AQ (Bishoff, Германия), и спектрофотометрическим детектором. Обработку хроматографической информации осуществляли при помощи программного обеспечения «Мультихром».
В работе использована субстанция анилокаина (ВФС 42-2946-97), синтезированная в ПГФА. В качестве биологической матрицы для приготовления калибровочных стандартов и контрольных образцов использовали плазму крови человека, полученную со станции переливания крови. Плазму хранили в сверхнизкотемпературном морозильном аппарате при температуре не выше 60°С.
Концентрированный раствор анилокаина с концентрацией 1 мг/мл был приготовлен путем растворения в метаноле. Рабочие растворы готовились путем соответствующих разведений исходных растворов водой. Калибровочные стандарты и контрольные образцы качества анилокаина готовили из рабочих растворов путем их разведения в бланковой плазме.
Для приготовления элюентов использовали воду, полученную в системе очистки воды Simplicity UV, кислоту трифторуксусную кислоту, ацетонитрил для хроматографии (сорт 0, Криохром, Санкт-Петербург).
Результаты исследований и их обсуждение
Проведенные ранее исследования по оптимизации условий изолирования анилокаина из плазмы крови показали, что в условиях модельного эксперимента максимальное излечение вещества (≈83%) наблюдается при использовании жидкость-жидкостной экстракции из щелочной среды после предварительного осаждения белков плазмы ацетонитрилом [5]. Для определения анилокаина в извлечениях использовали метод микроколоночной ВЭЖХ. Оптимальные результаты хроматографического разделения анилокаина и соэкстрактивных веществ были получены при градиентном элюировании в следующих условиях:
- элюент А – 0,1% раствор трифторуксусной кислоты, элюент В – ацетонитрил;
- режим элюирования градиентный: возрастание доли ацетонитрила с 10% до 70% за 20 минут, регенерация колонки 10% раствором ацетонитрила – 800 мкл;
- скорость потока элюента – 100 мкл/мин;
- детектирование – многоволновое (опорная длина волны - 210 нм, вспомогательные длины волн: 220, 230, 240, 250, 260, 280, 300 нм);
- температура термостата – 40°С;
- объем вводимой пробы – 20 мкл.
В данных условиях абсолютное время удерживания анилокаина составляет ≈ 9,5 мин.
Валидационную оценку биоаналитической методики проводили по следующим параметрам: специфичность, линейность, правильность, прецизионность, предел обнаружения и количественного определения, стабильность [4].
Специфичность разработанных условий была оценена путем сравнения хроматограмм экстрактов холостого образца плазмы и модельной смеси плазмы с известным содержанием аналита. Примеры хроматограмм приведены на рис.1-2.
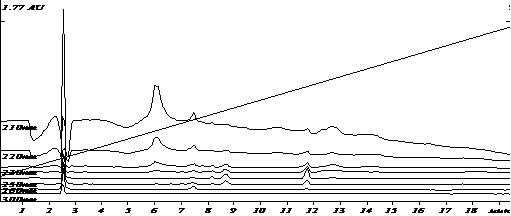
Рис.1. Хроматограмма извлечения из бланковой плазмы (холостой опыт)
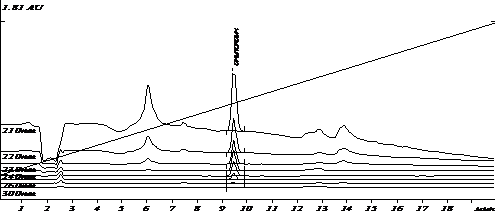
Рис.2. Хроматограмма извлечения из модельной смеси плазмы с содержанием анилокаина (500 нг/мл)
Анализ хроматограмм «холостого» опыта показал отсутствие пиков эндогенных веществ плазмы на времени выхода анилокаина. Коэффициент разделения пика анилокаина и соседних пиков составил не менее 5.
Для установления линейности методики были проанализированы 8 образцов модельных смесей плазмы с содержанием анилокаина от 42 до 2100 нг/мл. Результаты представлены на рис.3 и таблице 1.
Градуировочная зависимость описывается уравнением S =0,0024×С (S – площадь хроматографического пика, С – концентрация анилокаина в плазме, нг/мл), коэффициент корелляции R2 составил 0.9963. Предел количественного определения анилокаина в плазме по предложенной методике – 40 нг/мл.
Таблица 1
Отклонения концентраций калибровочных стандартов от фактических значений
|
С факт., нг/мл |
42 |
105 |
210 |
525 |
840 |
1050 |
1575 |
2100 |
|
С рассч., нг/мл |
37,5 |
112,5 |
205,0 |
508,3 |
862,5 |
1125,0 |
1500,3 |
2033,3 |
|
ε,% |
–10,7 |
7,1 |
–2,4 |
–3,2 |
2,7 |
7,1 |
–4,8 |
–3,2 |
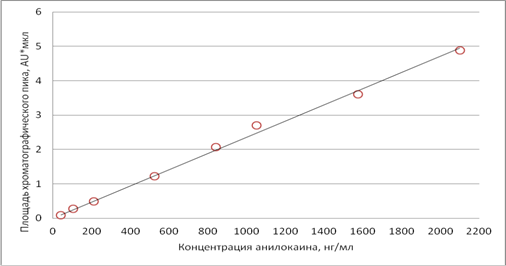
Рис.3. Зависимость площади пика от концентрации анилокаина (нг/мл) в калибровочных стандартах
Полученные отклонения концентраций калибровочных стандартов от фактических значений соответствуют современным требованиям, предъявляемым к биоаналитическим методикам [4].
Для оценки параметров методики «Прецизионность» и «Правильность» готовили по 6 модельных образцов плазмы на 5 уровнях концентраций анилокаина: 39; 117; 468; 936, 1404 нг/мл (контрольные образцы качества). Подготовку образцов для анализа осуществляли в соответствии с описанной методикой. Прецизионность и правильность методики оценивалась по величинам относительного стандартного отклонения (RSD, %) и относительной погрешности (ε,%) соответственно. Метрологические характеристики методики представлены в таблице 2. Полученные результаты контроля не превышают 15%, допускаемых для биоаналитических методик, что свидетельствует об отсутствии значимых систематических ошибок в результатах анализа [4].
Таблица 2
Прецизионность и правильность методики количественного определения анилокаина в плазме крови методом ВЭЖХ
|
Концентрация анилокаина в модельной смеси, нг/мл |
Найденная концентрация (нг/мл), |
SD |
RSD,% |
ε,% |
|
39 |
40,3 (44,6; 37,9; 40,1; 34,7; 42,8; 41,8) |
3,5 |
8,7 |
3,3 |
|
117 |
119,5 (122,4; 126,0; 107,1; 128,3; 110,9; 122,5) |
8,4 |
7,1 |
2,1 |
|
468 |
460,4 (485,5; 451,9; 434,2; 479,9; 444,8; 466,1) |
20,1 |
4,4 |
-1,6 |
|
936 |
906,8 (911,2; 896,4; 940,2; 903,3; 915,0; 874,9) |
16,3 |
1,8 |
-3,2 |
|
1404 |
1387,3 (1351,6; 1298,1; 1340,1; 1464,2; 1398,9; 1470,6) |
59,2 |
4,3 |
-1,2 |
Изучена стабильность контрольных образцов качества с концентрацией анилокаина 39 и 1404 нг/мл при замораживании/размораживании (3 цикла) и при хранении в течение 12 часов при комнатной температуре. Доказано, что величина относительной погрешности рассчитанных значений концентраций от фактических не превышает 15%.
Выводы:
1. Разработана методика количественного определения анилокаина в плазме крови методом микроколоночной высокоэффективной жидкостной хроматографии со спектрофотометрическим детектированием.
2. Валидационная оценка показала высокую чувствительность, точность и воспроизводимость методики.
3. Разработанная методика может быть использована для количественного определения анилокаина в плазме крови на этапе доклинических и клинических фармакокинетических исследований препаратов анилокаина.
Рецензенты:Малкова Т.Л., д.фарм.н., профессор, заведующий кафедрой токсикологической химии ГБОУ ВПО ПГФА Минздрава России, г. Пермь;
Ярыгина Т.И., д.фарм.н., профессор, профессор кафедры фармацевтической химии ФОО ГБОУ ВПО ПГФА Минздрава России, г. Пермь.
Библиографическая ссылка
Сабирзянов Д.Р., Карпенко Ю.Н. ВАЛИДАЦИОННАЯ ОЦЕНКА ХРОМАТОГРАФИЧЕСКОЙ МЕТОДИКИ ОПРЕДЕЛЕНИЯ АНИЛОКАИНА В ПЛАЗМЕ КРОВИ // Современные проблемы науки и образования. 2015. № 2-2. ;URL: https://science-education.ru/en/article/view?id=23077 (дата обращения: 01.07.2026).