



Введение
В результате поиска биологически активных соединений в ряду амидов
N-замещенных антраниловых кислот [4; 5] выявлено соединение, проявившее наибольший противовоспалительный эффект – амид N–аллилантраниловой кислоты (АNААК). В связи с рекомендацией к проведению доклинических испытаний нами проводятся исследования по разработке методов контроля качества и стандартизации субстанции и лекарственных форм этого соединения [1].
Исходя из схемы получения АNААК, посторонней (специфической) примесью в его субстанции может быть амид антраниловой кислоты (ААК).
Целью настоящей работы явилась разработка и валидация методики определения посторонней примеси ААК в субстанции АNААК методом обращённо-фазной ВЭЖХ.
Материалы и методы исследования
В исследованиях использовался высокоэффективный жидкостный хроматограф «Shimadzu LC Prominence» (Япония), оснащённый колонкой из нержавеющей стали (25 см×4,6 мм, сорбент Discovery® С18 с размером частиц 5 мкм) и диодноматричным детектором.
Для приготовления подвижных фаз использовали воду бидистиллированную, фосфорную кислоту концентрированную, трифторуксусную кислоту, ацетонитрил для хроматографии, метанол [3].
В работе использовали пять серий субстанции АNААК, синтезированные на кафедре фармацевтической химии ФОО ПГФА в 2008-2012 гг., ААК марки Lancaster Synthesis.
Результаты исследований и их обсуждение
Предварительный эксперимент показал, что оптимальным элюентом с точки зрения эффективного разделения аналитов, а также симметрии хроматографических пиков является подвижная фаза на основе ацетонитрила и 0,1%-ного раствора трифторуксусной кислоты в соотношении 30:70. УФ-спектры АNААК в 0,1 М растворе хлористоводородной кислоты характеризуются тремя максимумами поглощения 216 нм, 257 нм и 350 нм (Е1%1см соответственно 1520, 520 и 320) [2]. Длину волны детектирования (212 нм) выбирали, исходя из полученного спектра ААК в подвижной фазе (рис. 1). Хроматограмма раствора модельной смеси АNААК и ААК (по 200 мкг/мл) в элюенте представлена на рис. 2.
Разработанные условия хроматографического анализа:
- подвижная фаза: ацетонитрил – 0,1%-ный раствор трифторуксусной кислоты (30:70);
- скорость потока подвижной фазы – 1 мл/мин;
- длина волны детектирования – 212 нм;
- объем вводимой пробы – 20 мкл;
- температура термостата колонки – 40 °С.

Рис. 1. УФ-спектр ААК в подвижной фазе ацетонитрил – 0,1%-ный раствор трифторуксусной кислоты (30:70).
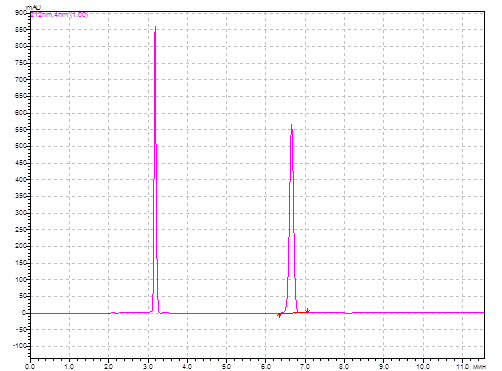
Рис. 2. Хроматограмма раствора модельной смеси АNААК и ААК (длина волны детектирования 212 нм).
Валидацию аналитической методики проводили по параметрам: специфичность, линейность, правильность, повторяемость (сходимость), предел обнаружения и предел количественного определения.
При подтверждении специфичности использовали модельную смесь субстанции АNААК (1 мг/мл) и примеси ААК (10 мкг/мл) в подвижной фазе (рис. 3).
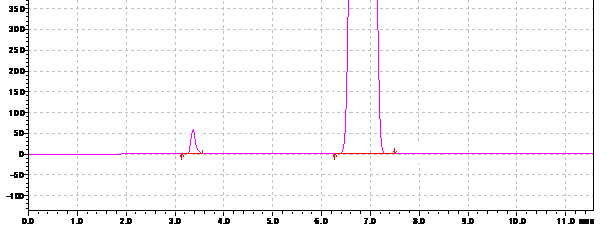
Рис. 3. Хроматограмма модельной смеси АNААК (1 мг/мл) и примеси ААК (10 мкг/мл) в подвижной фазе.
Идентификацию ААК и АNААК на хроматограмме осуществляли путем сопоставления времён удерживания аналитов и стандартных образцов. Пик ААК в разработанных условиях имеет время удерживания 3,20 ±0,05 мин, пик АNААК – 6,72±0,05 мин. Анализ «холостой» хроматограммы (растворителя образца) показал отсутствие мешающих посторонних пиков. Коэффициент разрешения (Rs) пика примеси и основного вещества составил 5,4, что свидетельствует о хорошем разделении (рекомендуемое значение Rs при определении содержания примесей больше 2). Коэффициент асимметрии пика ААК, характеризующий надежность определения границ пика, равен 1,1 (рекомендуемое значение – от 0,8 до 1,5). Относительное стандартное отклонение (RSD) площадей пиков ААК для трёх последовательных хроматограмм составило 1,92%.
Линейность методики определяли на пяти уровнях концентраций ААК в подвижной фазе (0,05; 0,1; 0,25; 0,35; 0,5% от содержания основного вещества – субстанции АNААК). Для этого в мерные колбы вместимостью 25 мл помещали раствор ААК в метаноле с концентрацией 53,5 мкг/мл (0,5, 1,0, 2,5, 3,5 и 5,0 мл) и доводили объем колб метанолом до метки. Коэффициент корреляции составил 0,999, что свидетельствует о линейности методики в выбранном диапазоне концентраций (рис. 4).
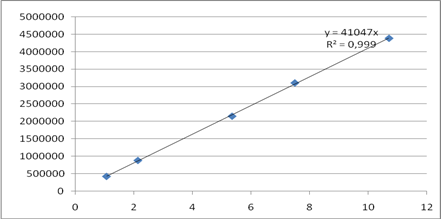
Рис. 4. Зависимость площади пика от концентрации ААК (мкг/мл) в модельных смесях.
Предел обнаружения ААК по предложенной методике составляет 0,5 мкг/мл (0,025% от содержания в субстанции АNААК), предел количественного определения 1 мкг/мл (0,05% от содержания в субстанции АNААК).
Оценку повторяемости методики проводили на модельных растворах ААК на четырех уровнях концентраций: 1, 2, 5 и 10 мкг/мл, что соответствует 0,05; 0,1; 0,25 и 0,5% от содержания в субстанции АNААК. Каждый из растворов готовился в соответствии с тестируемой методикой и хроматографировался не менее 3 раз.
Относительное стандартное отклонение не превышает 10,0%, что свидетельствует об удовлетворительной сходимости результатов на всех уровнях рассматриваемых концентраций (табл. 1).
Таблица 1 – Оценка повторяемости (сходимости) методики определения ААК
|
Содержание ААК в растворе, мкг/мл |
Найденное содержание ААК, мкг/мл |
Метрологические характеристики (P =0,95; n=6) |
||||
|
n |
|
SD |
RSD |
ΔX |
||
|
1 |
0,79; 0,87; 0,91; 0,78; 0,89;0,84 |
6 |
0,85 |
0,053 |
6,23 |
0,14 |
|
2 |
2,03; 1,93; 1,95; 2,04; 1,98; 1,95 |
6 |
1,98 |
0,046 |
2,32 |
0,12 |
|
5 |
4,84; 5,10; 4,90; 4,89; 5,06; 4,86 |
6 |
4,94 |
0,110 |
2,22 |
0,28 |
|
10 |
9,82; 9,88; 10,11; 10,07; 9,93; 9,78 |
6 |
9,93 |
0,133 |
1,34 |
0,34 |
При определении правильности методики (табл. 2) оценивали открываемость известного количества аналита (ААК), введенного в плацебо (субстанцию АNААК). Исследования проведены на трех уровнях содержания ААК в АNААК (0,05, 0,1 и 0,5%). По 0,05 г субстанции АNААК помещали в мерные колбы вместимостью 25 мл, растворяли в 10 мл метанола, добавляли в каждую колбу раствор ААК в метаноле с концентрацией 50 мкг/мл (0,5, 1,0, 5,0 мл) и доводили объем колб метанолом до метки.
Таблица 2 – Оценка правильности методики определения ААК
|
Содержание ААК в растворе, мкг/мл |
Открываемость (R), % |
Метрологические характеристики (P =0,95; n=6) |
||||
|
n |
|
SD |
RSD |
Δ |
||
|
1 |
92,4; 83,6; 102,0; 93,1; 88,2; 96,1 |
6 |
92,3 |
5,83 |
6,31 |
6,11 |
|
2 |
94,5; 103,5; 103,0; 97,0; 94,0; 98,5 |
6 |
98,42 |
4,09 |
4,15 |
4,29 |
|
10 |
98,8; 99,7; 102,1; 99,1; 101,4; 99,7 |
6 |
100,13 |
1,32 |
1,32 |
1,38 |
Анализ пяти серий субстанции АNААК показал, что содержание в них посторонней примеси ААК не превышает 0,1%.
Разработанная методика включена в проект ФС на субстанцию АNААК.
Выводы
1. Установлены условия определения примеси ААК в субстанции АNААК методом обращённо-фазной ВЭЖХ.
2. Валидация разработанной методики по параметрам специфичность, линейность, предел обнаружения и предел количественного определения, повторяемость (сходимость), правильность показала её приемлемость для определения 0,05-0,5% примеси ААК в субстанции АNААК.
Рецензенты:
Вихарева Елена Владимировна, доктор фармацевтических наук, доцент, заведующий кафедрой аналитической химии ГБОУ ВПО «ПГФА» Минздрава России, г. Пермь.
Гейн Владимир Леонидович, доктор химических наук, профессор, заведующий кафедрой физической и коллоидной химии ГБОУ ВПО «ПГФА» Минздрава России, г. Пермь.
Библиографическая ссылка
Карпенко Ю.Н., Басс С.М., Ярыгина Т.И. РАЗРАБОТКА И ВАЛИДАЦИЯ МЕТОДИКИ ОПРЕДЕЛЕНИЯ ПОСТОРОННЕЙ ПРИМЕСИ В СУБСТАНЦИИ АМИДА N–АЛЛИЛАНТРАНИЛОВОЙ КИСЛОТЫ // Современные проблемы науки и образования. 2013. № 1. ;URL: https://science-education.ru/ru/article/view?id=8511 (дата обращения: 04.08.2026).