


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
THE SYNTHESIS OF TRYPTAMINES AND THEIR HOMOLOGUES

Триптаминовые алкалоиды, их синтетические аналоги, а также эргоалкалоиды - производные лизергиновой кислоты - занимают заметное место в арсенале современных лекарственных средств. Такие препараты, как серотонин, мексамин, мелатонин, суматриптан, являются 5-замещенными триптаминами и достаточно продолжительное время используются в качестве регуляторов ЦНС [1].
Среди известных способов синтеза 5-замещенных триптаминов наибольшее распространение получили методы Абрамовича-Шапиро и Манске-Робинсона, ключевые стадии которых представлены на схемах 1 и 2 соответственно.
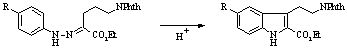
Схема 1
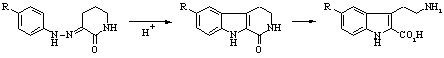
Схема 2
В обоих случаях получаются производные триптамина, имеющие в положении 2 индольного кольца карбоксильную группу, удаление которой на заключительной стадии синтеза требует жестких условий и в ряде случаев протекает с низким выходом.
Метод, предложенный Виноградом Л.Х. и Суворовым Н.Н. [2], исключает стадию декарбоксилирования. Этот метод предлагает использование в качестве исходных соединений 4-замещенных фенилгидразинов и g-фталимидомасляного альдегида. Реакция протекает в мягких условиях с высокими выходами по схеме 3.
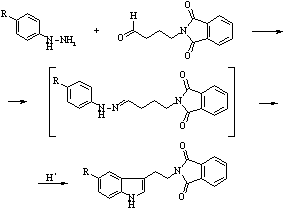
Схема 3
В литературе описано два способа получения g-фталимидомасляного альдегида. Первый в качестве ключевой стадии предусматривает восстановление хлорангидрида соответствующей кислоты по методу Розенмунда [3]. Второй включает стадии оксосинтеза [4], каталитического гидрирования и защиту аминогруппы с использованием N-карбметоксифталимида.
У всех выше перечисленных схем имеется ряд недостатков: все они требуют использования дорогостоящих катализаторов, в ряде случаев - и специальной аппаратуры; чувствительны к следам воды.
Поэтому разработка удобных препаративных методов синтеза w-фталимидоалкановых альдегидов, триптаминов и гомотриптаминов на их основе является в настоящее время задачей актуальной.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Общая методика получения w-фталимидоалкановых альдегидов
В круглодонную колбу емкостью 0,5 л помещают 0,020 моль a-окси-e-фталимидо-алкановой кислоты, 200 мл воды и 2,02 г (0,024 моль) NaHCO3, после полного растворения твердой фазы добавляют 5,47 г (0,024 моль) H5IO6. Реакционную смесь нагревают на водяной бане при 45° при постоянном перемешивании в течение 24 часов. Контроль за ходом реакции осуществляют методом ТСХ. После завершения реакции растворитель отгоняют в вакууме досуха. К остатку приливают 100 мл бензола, размешивают 5-10 минут и фильтруют. Осадок еще раз промывают 50 мл бензола. Бензольный раствор переносят в делительную воронку, промывают 25 мл воды, затем 25 мл 5%-ного раствора NaНCO3 и снова 25 мл воды. Раствор сушат над MgSO4 в течение 12 часов и растворитель упаривают под вакуумом.
Получают 90% d-фталимидоалканового альдегида в виде светло-желтого масла.
Общая методика получения триптаминов и гомотриптаминов
В круглодонную колбу емкостью 0,25 л помещают 4,6 ммоль g-фталимидоалканового альдегида и 0,42 г (3,8 ммоль) фенилгидразина. Смесь нагревают на водяной бане при температуре 50°С при постоянном перемешивании в течение часа. Затем приливают 100 мл 8% трифторуксусной кислоты, пускают в ход механическую мешалку и нагревают на кипящей водяной бане. Окончание реакции контролируют методом ТСХ.
По окончании реакции катализатор декантируют, реакционную массу промывают 100 мл воды, нейтрализуют 25% раствором аммиака (около 6 мл), и снова 100 мл воды. Полученный продукт высушивают азеотропно с бензолом. Бензольный раствор упаривают досуха, а полученный продукт перекристаллизовывают из метанола. Выходы полученных соединений представлены в таблице 3.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Как сообщалось ранее [5] фталимидомасляный альдегид (схема 4) может быть получен окислением соответствующей оксикислоты периодатом натрия по методу Янука-Сареля-Каца [6] (схема 4).
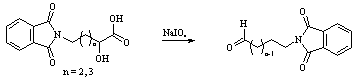
Схема 4
При проведении реакции в условиях, описанных авторами, целевые альдегиды образуются с выходом примерно 80%.
При отработке метода было установлено, что лучшие результаты дает окисление натриевой соли соответствующей окси кислоты периодной кислотой в воде. Выход целевого альдегида составил около 90%, а содержание основного вещества, определенное оксимным титрованием, составляло около 99%.
Исходные оксикислоты [7] были получены, исходя из соответствующих w-аминокислот, которые образуются с высоким выходом при гидролизе лактамов (схема 5).
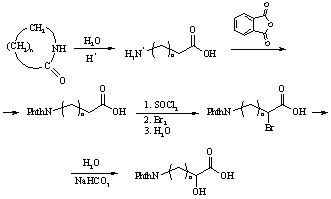
Схема 5
На основе отработанного метода получения фталимидоалканового альдегида был проведен синтез ряда 5-замещенных фталимидотриптаминов.
Известно, что успех проведения синтеза индолов, не имеющих заместителей в положении 2 по Э. Фишеру, зависит от целого ряда факторов: растворителя, в котором проводится циклизация, кислотного катализатора, времени и температуры реакции, наличия заместителей в бензольном кольце и положении 3 индольного цикла [8, 9].
Синтез N-фталил- триптаминов и гомотриптаминов был осуществлен нами по методу, предложенному Суворовым и Виноградом [2]. В качестве исходных соединений использовался 4-замещенный фенилгидразин и фталимидоалкановый альдегид [5].
Гидразоны соответствующих альдегидов могут использоваться как исходные вещества или получаются непосредственно перед реакцией в том же растворителе и после прибавления кислого катализатора циклизуются в индол. Нами был выбран второй вариант, как наиболее простой и не сопряженный с потерями вещества при выделении.
В качестве растворителей нами были проверены спирты (метиловый и этиловый) и уксусная кислота. Кислотные катализаторы приведены в таблице 1.
Таблица 1. Условия получения N-фталилгомотриптамина
|
Растворитель |
Катализатор |
Условия реакции |
Результат |
|
1. Метанол |
¾ |
Нагревание 12 ч |
¾ |
|
2. Метанол |
H2SO4 |
Нагревание 6 ч |
¾ |
|
3. Уксусная кислота |
¾ |
Нагревание 6 ч |
¾ |
|
4. Этанол |
ZnCl2 |
Нагревание 10 ч |
¾ |
|
5. Этанол |
HCl |
Нагревание 4 ч |
¾ |
|
6. Этанол |
Уксусная кислота |
Нагревание 10 ч |
¾ |
|
7. Этанол |
H3PO4 |
Нагревание 5 ч |
60% |
Единственной комбинацией «растворитель - катализатор» является комбинация этиловый спирт-фосфорная кислота (85%) при температуре кипения смеси.
При уточнении условий циклизации мы установили, что соотношение исходный гидразин : H3PO4 должно быть 1 : 3-4. При меньшем соотношении реакция (по данным ТСХ) не заканчивается даже при кипячении в течение 72 часов.
Отработанным методом был получен ряд триптаминов и гомотриптаминов с достаточно высокими выходами, но полученные продукты представляли из себя маслообразные продукты. Выделение продукта перекристаллизацией из различных растворителей не позволило получить желаемый продукт в виде индивидуального соединения. Триптамины были выделены хроматографией на колонке с окисью алюминия. После хроматографической очистки выходы целевых соединений не превышали 30%.
При получении гидразона без растворителя и использовании в качестве катализатора циклизации водных растворов кислот (таблица 2) в ряде случаев выходы целевых соединений значительно возросли, но очистка соединений требовала многократной перекристаллизации.
Лучшие результаты были нами получены при использовании для циклизации фенилгидразона 8% водного раствора трифторуксусной кислоты. Гидразон получали реакцией гидразина с альдегидом без растворителя. Соотношение гидразин : альдегид 1 : 1,2. N-Фталимидотриптамин был получен нами с выходом 80%. После кристаллизации из бензола, а затем из метанола температура плавления составила 165-166°С.
Таблица 2. Условия получения N-фталимидотриптамина
|
Растворитель |
Катализатор,% |
Условия реакции |
III, Выход% |
|
Этанол |
H3PO4 (85) |
Нагревание 5 ч |
60 |
|
Вода |
H3PO4 (8) |
Нагревание 10 ч |
Следы |
|
Вода |
H2SO4 (4) |
Нагревание 6 ч |
69 |
|
Вода |
HCl (8) |
Нагревание 12 ч |
Следы |
|
Вода |
CF3CO2H (8) |
Нагревание 1 ч |
80 |
|
Вода |
CF3CO2H (8) |
Нагревание 1,5 ч |
79,5 |
|
Вода |
CF3CO2H (8) |
Нагревание 2 ч |
81 |
При использовании в качестве катализатора 8% трифторуксусной кислоты были получены N-фталимидотриптамин, N-фталимидогомотриптамин, 5-бром-N-фталимидотриптамин, 5-бром-N-фталимидогомотриптамин, 5-метил-N-фталимидотриптамин, 5-метил-N-фталимидогомотриптамин (таблица 3).
Таблица 3
|
№ п/п |
Соединение |
Выход,% |
Тпл, °С |
ИК спектр,n, см-1 |
|
1 |
N-фталимидотриптамин |
80 |
165-166 |
3429 (NH); 1770 (CO); 1712 (CO); 721 (-CH2-) |
|
2 |
N-фталимидо-гомотриптамин |
69 |
128-130 |
3340 (NH); 1770 (CO); 1711 (CO); 720 (-CH2-) |
|
3 |
5-бром-N-фталимидо-триптамин |
65 |
211-212 |
3329 (NH); 1770 (CO); 1710 (CO); 719 (-CH2-) |
|
4 |
5-бром-N-фталимидо-гомотриптамин |
57 |
200-203 |
3406 (NH); 1766 (CO); 1720 (CO); 718 (-CH2-) |
|
5 |
5-метил-N-фталимидо-триптамин |
62 |
175-179 |
3395 (NH); 1767 (CO); 1720 (CO); 721 (-CH2-); 1396 (-CH3) |
|
6 |
5-метил-N-фталимидо-гомотриптамин |
59 |
163-165 |
3409 (NH); 1770 (CO); 1716 (CO); 721 (-CH2-); 1396 (-CH3) |
Образцы не давали депрессии с заведомыми образцами, синтезированными по методу Манске-Робинсона на кафедре Органической химии РХТУ им. Менделеева и любезно предоставленными нам к.х.н. Е.Н. Гордеевым. ИК-спектры соединений идентичны.
ЗАКЛЮЧЕНИЕ
Достоинством отработанного синтеза являются:
- 1) доступность сырья для получения альдегидов,
- 2) сравнительная простота и хорошая воспроизводимость,
- 3) высокие выходы,
- 4) широкая применимость.
Было установлено, что при увеличении загрузки в 3 и 9 раз выход целевого продукта не претерпевает значительных изменений, а расхождения находятся в допустимых пределах.
СПИСОК ЛИТЕРАТУРЫ:
- 1. Машковский М.Д. Лекарственные средства. - 15-е изд., перераб., испр. и доп. - М.: ООО «Издательство Новая Волна», 2006. - 1200 с.
- 2. Виноград Л.Х., Суворов Н.Н. //Химия гетероциклических соединений. 1984. № 9. С. 1206.
- 3. Balenovic K., Jambresic I., Furic I. //Journal Organic Chemistry. 1952. Vol. 17. P. 1459.
- 4. Кацнельсон М.Г, Дельник В.Б., Кагна С.Ш., Кашина В.В, Леенсон Е.И. //Нефтехимия. 1982. Т. XXII. №5. С.683.
- 5. Sevodin P.V. Mitrofanov R.Y., Vereshchagin A.L., Sevodin V.P. //International conference on natural products and physiologically active substances (ICNPAS-98): Novosibirsk, Russia November 30 - December 6. 1998. P. 160.
- 6. Yanuka Y., Katz R., Sarel S. //Tetrahedron Letters. 1968. № 14. P. 1725.
- 7. Севодин П.В. Митрофанов Р.Ю., Верещагин А.Л., Севодин В.П. //Материалы Всероссийской научно-практ. конф. «Прикладные аспекты совершенствования химических технологий и материалов». В 2-х частях. Часть 1. - Бийск: АлтГТУ, 1998, с. 56.
- 8. Deorha D.S., Joshi S.S. //Journal Organic Chemistry. 1961. Vol. 26. № 9. P. 3527.
- 9. Desaty D., Keglevic D. //Croatica Chemica Acta. 1965. Vol. 37. P. 25.
Библиографическая ссылка
Митрофанов Р.Ю, Севодин В.П СИНТЕЗ ТРИПТАМИНОВ И ИХ ГОМОЛОГОВ ИЗ -ФТАЛИМИДО-АЛКАНОВЫХ КИСЛОТ // Современные проблемы науки и образования. 2007. № 6-1. ;URL: https://science-education.ru/en/article/view?id=758 (дата обращения: 14.07.2026).