


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
INFLAMMATION AS A PATHOGENESIS FACTOR AND A TOOL FOR EARLY DIAGNOSIS OF CHRONIC KIDNEY DISEASE IN PATIENTS WITH TYPE I DIABETES MELLITUS

Введение
В настоящее время в мире насчитывается более полумиллиарда человек с сахарным диабетом (СД), к 2045 году число таких больных достигнет 630 миллионов [1]. Повреждение почек при СД первоначально обозначалось как «диабетическая нефропатия», а с 2007 года введено понятие «диабетическая болезнь почек» (ДБП). ДБП является одним из самых тяжелых осложнений СД и возникает примерно у трети больных с СД 1-го типа (СД1), приводит к возникновению терминальной стадии хронической болезни почек (ХБП) примерно у половины пациентов с СД. ДБП является шестой по распространенности причиной инвалидности и четвертой причиной смерти в мире [2]. В связи с этим комплексная профилактика, ранняя диагностика и разработка новых терапевтических стратегий имеют решающее значение для снижения прогрессирования ХБП при СД и связанной с этим смертности. Для диагностики ДБП традиционно используют такие критерии, как прогрессирующее снижение расчетной величины скорости клубочковой фильтрации (рСКФ), увеличение альбуминурии и отношения альбумина к креатинину в моче. Обычно ДБП начинается с увеличения рСКФ и транзиторной микроальбуминурии, затем снижается рСКФ, микроальбуминурия становится постоянной, нарастающая протеинурия является неблагоприятным прогностическим фактором ухудшения функции почек. Однако у некоторых больных с СД ХБП возникает без протеинурии, а вариабельность рСКФ не позволяет эффективно выявлять поражения почек при СД на начальных этапах [3].
В патогенезе ДБП участвуют множество факторов, включая гипергликемический профиль плазмы, накопление конечных продуктов гликирования (AGE), гемодинамические изменения, окислительный стресс, эндотелиальную дисфункцию, воспаление, фиброз и др. [4]. Воспаление занимает центральную позицию в патогенезе поражения почек при СД, прогрессировании ДБП, является источником многочисленных медиаторов, продуктов обмена, повреждения клеточных и внеклеточных структур, большинство из которых попадают в кровь и могут выступать в роли перспективных биомаркеров для раннего выявления заболевания и его прогрессирования, оценки эффективности терапевтических и профилактических мероприятий при ДБП. Контроль гликемии, применение ингибиторов ренин-ангиотензин-альдостероновой системы и прочих средств стабилизации гемодинамики при СД не могут полностью ингибировать прогрессирование ДБП. В качестве перспективных средств патогенетической терапии способны выступать регуляторы воспалительного процесса [5].
Цель исследования – провести критический анализ данных, посвященных роли воспалительного процесса в патогенезе и ранней диагностике ХБП у больных с СД1, в источниках, индексированных в Pubmed и Российском индексе научного цитирования.
Материалы и методы исследования
Для проведения систематического обзора в базах данных Pubmed, Российского индекса научного цитирования произведен поиск соответствующих исследований за 2019–2024 гг. по ключевым словам: «сахарный диабет первого типа», «хроническая болезнь почек», «диабетическая болезнь почек», «патогенез», «маркеры воспаления», «биомаркеры». Исключения из исследования: тезисы без полных текстов, источники, дублирующиеся или не относящиеся к рассматриваемой проблеме. Извлечение данных из каждого включенного источника: авторы, год публикации, план и результаты исследования. Стратегия поиска выявила 325 источников из PubMed и 20 из Российского индекса научного цитирования. После исключения дублирующих и нерелевантных записей в статью включены 49 исследований и проведен анализ их результатов. Подготовка и выполнение литературного обзора были основаны на современных принципах и порядке проведения систематических обзоров [6].
Результаты исследования и их обсуждение
Индукция воспаления при СД1. Патогенез воспаления при СД является многофакторным. Ключевым пусковым фактором служит гипергликемия – облигатный симптом любой формы СД. Прямое флогогенное действие глюкозы реализуется за счет увеличения осмотического давления плазмы, ОЦК, перфузии почек, что приводит к увеличению СКФ на начальных этапах СД и одновременно к гемодинамическому удару, повышению внутриканальцевого давления и повреждению клеток клубочка и канальцев. Кроме того, повышенный уровень глюкозы стимулирует синтез нефроэпителиоцитами провоспалительных факторов, включая молекулу межклеточной адгезии-1 (ICAM-1), хемоаттрактрантный моноцитарный белок-1 (MCP-1) и иные хемокины, остеопонтин. Эти провоспалительные факторы привлекают моноциты, нейтрофилы, лимфоциты, приводят к лейкоцитарной инфильтрации в межканальцевый интерстиций. Макрофаги и эпителиоциты, экспрессирующие CD40 и CD40L, через сигнальные каскады фосфоинозитид-3-киназы, ядерный фактор каппа-B (NF-κB), внеклеточные сигнальные киназы ERK (extracellular signal-regulated kinases) участвуют в синтезе провоспалительных и профиброгенных медиаторов [7]. Гипергликемия через активацию протеинкиназы C приводит к повышению экспрессии на эпителиоцитах канальцев Toll-подобного рецептора-4 (TLR-4) и высвобождению его лиганда HMGB-1, что потенцирует активацию NF-κB и синтез IL-6, MCP-1 и иных провоспалительных цитокинов [8]. Аналогичным образом TLR-2 может участвовать в синтезе провоспалительных цитокинов при СД через митоген-активированную протеинкиназу (MAPK), P38 и пути ERK, что сопровождается активацией NF-κB, инфильтрацией макрофагами тканей почек и синтезом эндогенных лигандов TLR, таких как белок теплового шока-70 (HSP-70) и HMGB1. Гипергликемия вызывает гликозилирование белков системы комплемента, включая C3, C3a, C5a, C5b–9, что приводит к его избыточной активации и участию в повреждении тканей почек, потенцированию хемотаксиса воспалительных клеток [9]. Под влиянием гипергликемии, дополнительных профиброгенных факторов, прежде всего IL-1β, TGF-β1, эпителиоциты почечных канальцев подвергаются фенотипической эпителиально-мезенхимальной трансформации в миофибробласты, синтезируют компоненты внеклеточного матрикса, включая ламинин, фибронектин и коллаген IV типа, и усугубляют повреждение почек [10]. TGF-β1 препятствует экспрессии и активности матричных металлопротеиназ, которые имеют решающее значение для деградации компонентов внеклеточного матрикса, оказывая дополнительное профиброгенное действие.
Опосредованные, независимые от глюкозы флогогенные механизмы при СД реализуются за счет эффектов конечных продуктов гликирования (AGE – advanced glycation end-products), липотоксичности и окислительного стресса (ОС) [11]. AGE образуются в результате неферментативных реакций между белками, липидами и углеводами, в частности глюкозой. Эпителиоциты проксимального канальца нефрона, в избытке реабсорбирующие AGE, синтезируют IL-8, растворимый ICAM-1, что способствует инфильтрации тубулоинтерстициального пространства лейкоцитами. AGE активируют CD4+ и CD8+ Т-лимфоцитов, которые проникают в почки и синтезируют IFN-γ и TNF-α [12]. Запуск воспалительного процесса, ОС приводят к нарушению структуры и функции гломерулярного барьера, повышению его проницаемости, появлению альбуминурии. Кроме этого, AGE инициирует активацию пути протеинкиназа С (PKC), индуцируя экспрессию ICAM-1, молекулу межклеточной адгезии сосудов-1 (VCAM-1), MCP-1. При связывании со своими рецепторами AGE повышают синтез TGF-β1, активируют почечные фибробласты, синтез коллагена [13]. Опосредованно через сигнальный путь ERK/p38-Smad3 AGE повышает секрецию эпидермального фактора роста, фактора роста соединительной ткани, потенцируя тубулоинтерстициальный фиброз. Несмотря на то что в норме эпителиоциты канальцев нефрона предпочитают в качестве энергоносителей жирные кислоты, а не глюкозу, в условиях дефицита инсулина при СД1 происходит избыточное образование и окисление жирных кислот, формируется липотоксичный эффект на почечную ткань [14]. В условиях гипергликемии на клетках канальцев нефрона повышается экспрессия CD36, отвечающего за перенос длинноцепочечных жирных кислот, что приводит к накоплению в клетках липидов, повышению синтеза холестерина и образованию пенистых клеток, что способствует прогрессированию альбуминурии и усугубляет повреждение почек [15].
Одним из индукторов и факторов поддержания воспаления в почках при СД1 выступает ОС [1, 6]. ОС формируется при СД за счет гипергликемии, активации полиолового и гексозаминового путей метаболизма глюкозы, образования AGE, активации НАДФН-оксидазы, миелопероксидазы, ксантиноксидазы, NO-синтазы и иных прооксидантных ферментов, что приводит к избыточной генерации активных форм кислорода (АФК) и азота. Кроме этого, имеет значение снижение синтеза и активности, в том числе за счет полиморфизма генов и эпигенетических изменений, ферментов антиокислительной защиты – супероксиддисмутазы, каталазы, глутатионпероксидазы, глутатионредуктазы, гемоксигеназы-1 и др. С одной стороны, ОС вызывает окислительную модификацию липидов, белков, нуклеиновых кислот и, как следствие, повреждение клеток и компонентов интерстиция в почечной ткани, что запускает воспалительный процесс. При этом повреждение компонентов митохондрий потенцирует ОС и замыкает порочный круг. С другой стороны, окислительное повреждение активирует воспалительные пути сигнализации: митоген-активированные протеинкиназы (MAPK), семейство факторов транскрипции, контролирующих экспрессию генов иммунного ответа, апоптоза и клеточного цикла (NF-κB), семейство ассоциированных с рецепторами киназ и активаторов транскрипции, участвующих в передаче сигнала (JAK-STAT), а также активирует иммунные клетки и способствует синтезу и секреции провоспалительных цитокинов. Как следствие, ОС, митохондриальная дисфункция, воспаление и гипергликемия при СД создают порочные круги, которые ускоряет прогрессирование ДБП [16].
Роль воспаления в патогенезе и прогрессировании диабетической нефропатии при СД1. Основой для реализации воспалительного процесса при ДБП служат клубочки и канальцы нефрона, сосудистая сеть и интерстиций. Морфологические и функциональные изменения на уровне клубочков и канальцев нефрона включают расширение мезангиального пространства, гломеруросклероз, утолщение клубочковой базальной мембраны, альбуминурию, тубулоинтерстициальный фиброз и прогрессирующее снижение функции почек [4]. Первичная и вторичная альтерация тканей почек при воспалении, ОС, увеличение синтеза профибротических факторов роста, включая фактор роста соединительной ткани, TGF-β, активируют продукцию фибробластами и миофибробластами внеклеточного матрикса, в том числе коллагенов I, III и IV типов, фибронектина, приводят к формированию фиброза и, в конечном итоге, гломерулосклероза. Особое значение придается эпителиально-мезенхимному и эндотелиально-мезенхимальному переходам, то есть трансформации эпителиальных и эндотелиальных клеток в миофибробласты [17].
Индукторами воспалительного процесса при СД1 выступают описанные выше эффекты гипергликемии, AGE, липидные метаболиты и липотоксичность, АФК и ОС, а также высвобождаемые при повреждении клеток АТФ, ДНК, РНК, ураты и иные факторы, которые взаимодействуют с паттерн-распознающими рецепторами, прежде всего и наиболее изученными TLR-2 и TLR-4, на поверхности иммунокомпетентных клеток, эпителиальных клеток нефрона, эндотелиоцитах, подоцитах и мезангиальных клеток, а также цитоплазматическими Nod-подобными рецепторами (NLR), в частности NLRP3 (рисунок). Активация TLR и NLR обеспечивается взаимодействием с молекулярными паттернами, связанными с повреждением (DAMP, damage-associated molecular pattern) и в меньшей степени – молекулярными паттернами, связанными с патогенами (PAMP, pathogen-associated molecular patterns). Экспрессия TLR-2 и TLR-4 положительно коррелирует с инфильтрацией интерстиция почек макрофагами, концентрацией HbA1C в крови, отрицательно коррелирует с рСКФ [18]. TLR2- и TLR4-зависимая внутриклеточная сигнализация при участии NF-κB опосредует синтез хемокинов и провоспалительных цитокинов при СД1. Установлена роль при СД1 таких лигандов TLR-2 и TLR-4, как эндотоксины, HSP60 и HMGB1, дополнительным механизмом активации TLR-4 выступает ангиотензин II. Образование комплекса инфламмасомы NLRP3 связано с активацией каспазы-1, каспазы-5 и секрецией IL-1β и IL-18, участвующих в патогенезе ДБП, что представляет интерес при разработке перспективных терапевтических стратегий [19]. Формирование NLRP3 инфламмасом имеет значение в прогрессировании ДБП через воздействие на клетки клубочкового аппарата нефрона, подоциты и клетки канальцев, повреждение клубочковой базальной мембраны, особенно гибель подоцитов, поддерживающих структурную целостность барьера, и является ранним признаком ДБП. Появление NLRP3 инфламмасом приводит к альбуминурии и протеинурии [20].
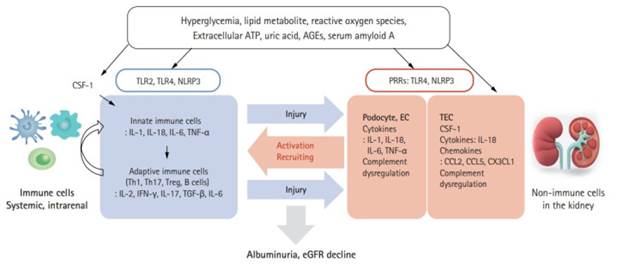
Индукция воспаления и механизмы его реализации при ДБП [21]
Реализация воспалительного ответа при ДБП во многом связана с функциональной активностью клеток врожденного и адаптивного иммунитета. Так, выраженность диабетической нефропатии коррелирует с инфильтрацией почечной ткани при СД макрофагами и дендритными клетками, при этом способствующим фактором выступает экспрессия молекул адгезии, таких как ICAM-1, VCAM-1, и факторов хемотаксиса, в частности MCP-1 [22]. При ДБП в клинических и экспериментальных условиях наблюдается увеличение количества внеклеточных ловушек нейтрофилов (NET) в почечной ткани, что было ассоциировано с повреждением и гибелью путем пироптоза эндотелиоцитов капилляров клубочка нефрона. С NET-индуцированным пироптозом клеток гломерулярного фильтра связывают повышение его проницаемости при СД1 и формирование альбуминурии [23]. Меньшее значение в патогенезе ДБП при СД1 имеет инфильтрация почечной ткани лимфоцитами: установлена роль увеличения клеток Th1 и Th17 и уменьшения Treg в активации Th1-зависимых и подавлении Th2-зависимых реакций [24].
Определенную роль в повреждении почек при СД1, инициировании и прогрессировании ДБП играет система комплемента. Активация системы комплемента происходит по альтернативному и лектин-зависимому путям в ответ на гликированные белки и в результате дисфункции гликированных белков-регуляторов активности комплемента, в частности CD59 [25]. Установлены повышенная экспрессия компонентов С5 и С3 в почках, ассоциированная с их дисфункцией при СД, и положительный эффект применения ингибиторов C5aR1 [26]. Повышенный уровень маннозосвязывающих лектинов (MASP-1, MASP-2 и MASP-3) в плазме на ранних стадиях СД1 связан с микро- или макроальбуминурией, а также с повышенной смертностью [27].
Большое значение при воспалении у больных с ДБП имеют цитокины, в том числе IL-1β, IL-6, IL-17, IL-18, TNF-α, INF-γ, TGF-β, MCP-1 [22]. Обнаружена прямая корреляция между прогрессированием ДБП, альбуминурией и увеличением концентрации в сыворотке, а также в почечной ткани провоспалительных цитокинов, VCAM-1, ICAM-1 [16]. В последнее время большое значение придается избыточному синтезу и эффектам MCP-1, не только связанным с хемотаксисом и рекрутингом моноцитов и макрофагов в почечную паренхиму, но также в индукции фиброзных реакций и гемодинамических изменениях, ведущих к альбуминурии [28]. TNF-α участвует в индукции воспалительного ответа, апоптоза, регуляции иммунного ответа, увеличении проницаемости сосудов, формировании ОС. Уровень TNF-α в клетках клубочков и канальцев нефрона напрямую и независимо связан с альбуминурией у больных с СД1. В когорте пациентов с СД1 с нормо- и микроальбуминурией сывороточные растворимые рецепторы TNF-α (TNFR1 и TNFR2) были тесно связаны со снижением рСКФ независимо от альбуминурии [21]. IL-1β индуцирует синтез простагландина Е и активацию фосфолипазы A2 в мезангиальных клетках, что имеет значение в формировании гломерулярной гиперперфузии, повышении проницаемости и адгезивности эндотелия. IL-6 участвует в инфильтрации нейтрофилами тубулоинтерстиция, гипертрофии подоцитов и утолщении клубочковой базальной мембраны, его уровень коррелирует с альбуминурией. IL-18 опосредует апоптоз клеток почек, синтез и высвобождение IL-1β, TNF-α, INF-γ и экспрессию молекул адгезии ICAM-1. В настоящее время пересматривается однозначно негативная роль IL-17A при ДБП, его эффекты не ограничиваются провоспалительным и профиброгенным действиями, связаны также с регуляцией аутофагии и сменой фенотипа макрофагов [29].
Провоспалительные цитокины (IL-6, TNF-α, INF-γ) наряду с описанными выше событиями участия в реализации воспаления в почках при СД1 индуцируют синтез в гепатоцитах группы белков плейотропного действия, концентрация которых изменяется в сыворотке, – реактантов острой фазы (РОФ). Накоплены сведения об участии РОФ в патогенезе диабетической нефропатии при СД1. С-реактивный белок (С-РБ) из семейства пентраксинов участвует в активации системы комплемента, секреторной функции нейтрофилов, фенотипической перестройке макрофагов в М1, зависимо от экспрессии C3aR ингибирует C3a-индуцированную аутофагию подоцитов, что имеет значение в прогрессировании диабетической нефропатии [30]. Различные изоформы С-РБ могут как участвовать в повреждении тканей почек, усугублять воспаление и фиброз (пентамер С-РБ) через активацию путей NF-κB, TGF-β/Smad3, Wnt/β-catenin, так и проявлять противовоспалительные свойства (мономер С-РБ) через блокаду пути C3a/C3aR [31]. Концентрация в сыворотке С-РБ при ДБП коррелирует с экспрессией дипептидилпептидазы-4 (DPP4) – еще одного участника воспалительного процесса в почках при СД [32]. Дефицит СРБ ингибирует диабетическое повреждение почек у нокаутированных по гену С-РБ крыс [30].
Церулоплазмин (ЦП) – РОФ второго эшелона, медьсодержащий белок альфа-2-глобулиновой фракции плазмы с выраженной плейотропной активностью, участвует в регуляции воспаления, редокс-статуса, обмене меди и железа, окислении ЛПНП и иных процессах [33]. Противовоспалительные свойства ЦП реализуются благодаря его взаимодействию с лактоферрином и миелопероксидазой, которые ограничивают ОС [34]. При СД1 наблюдаются нарушения метаболизма меди и повышенная концентрация ЦП в сыворотке [35]. Ряд исследователей полагают, что концентрация в сыворотке ЦП является более чувствительным маркером ДБП, чем концентрация высокочувствительного С-РБ (вчС-РБ) [36]. Метаболизм углеводов и иных веществ при СД существенно нарушается при дефиците и избытке меди, в том числе формируется ОС за счет снижения синтеза Cu, Zn-супероксиддисмутазы, имеющий значение в патогенезе ДБП; есть данные, указывающие на повышение уровня меди в сыворотке при СД1. Повышение концентрации меди в почке и в крови за счет как повышения синтеза ЦП в ходе ответа острой фазы, так и снижения клиренса меди в условиях падения СКФ при ДБП приводит к отложению меди и повреждению клеток почечных канальцев, что усугубляет нарушение функции почек при СД [37]. Кроме этого, при СД в условиях прогрессирующей почечной дисфункции выведение меди с мочой обусловлено диссоциацией комплексов «альбумин – медь» и «церулоплазмин – медь», фильтрующихся через поврежденную гломерулярную мембрану.
Перспективные биомаркеры воспаления как инструменты ранней диагностики ДБП в клинической практике. Ранняя диагностика ДБП важна для построения индивидуальной терапевтической траектории при СД1 с целью замедления прогрессирования снижения функции почек и предотвращения терминальной стадии ХБП, существенно ограничивающей качество жизни и требующей проведения заместительной почечной терапии или трансплантации почки. С учетом патогенеза ДБП, включающего наряду с факторами воспалительного процесса метаболические и локальные гемодинамические изменения при СД1, используют традиционные биомаркеры, такие как концентрация креатинина в сыворотке, рСКФ, альбуминурия, цистатин С [38]. Перспективные биомаркеры воспаления при ДБП у больных с СД1 включают гетерогенную группу факторов: молекулу повреждения почки-1 (KIM-1), нейтрофильный желатиназно-ассоциированный липокалин (НЖАЛ), цитокины и хемокины (TNF-α и его рецепторы, MCP-1), количество в крови лимфоцитов, нейтрофилов, отношение лимфоциты/нейтрофилы, РОФ С-РБ и ЦП, маркеры ОС.
KIM-1 – гликопротеин повреждения клеток проксимальных канальцев, его концентрация в сыворотке повышается при ДБП, независимо связана с альбуминурией, уровнем в сыворотке креатинина и HbA1c. KIM-1 отражает выраженность апоптоза эпителиоцитов канальцев нефрона, поглощение жирных кислот на уровне проксимального извитого канальца, инициируя воспаление и фиброз. Чувствительность и специфичность определения KIM-1 составляют 93,8% и 88,5% соответственно [39]. Диагностическая ценность KIM-1 подтверждена при экспериментальном моделировании СД1 [40]. НЖАЛ – компонент нейтрофильных гранул, повышение его концентрации в сыворотке и в моче отражает повреждение эпителиальных клеток проксимальных канальцев нефрона и снижение реабсорбции свободно фильтруемого НЖАЛ, обратно коррелирует с рСКФ и гистологическими маркерами повреждения почек, но не с HbA1c. Чувствительность и специфичность определения НЖАЛ составляют 94% и 90% соответственно. Кроме этого, НЖАЛ выступает в качестве чувствительного биомаркера острого повреждения почек при СД1 [41]. В целом, НЖАЛ является перспективным биомаркером для раннего выявления, мониторинга, стратификации риска и оценки реакции на лечение при ДБП [42].
TNF-α и его рецепторы TNFR1, TNFR2, MCP-1 – маркеры системного воспаления при ДБП. Повышение концентрации TNF-α в сыворотке и в моче ассоциировано с альбуминурией [43]. TNFR1 и TNFR2 являются сильными независимыми предикторами снижения функции почек, повышенные концентрации TNFR1 и TNFR2 (TNFR2 имеет самый высокий риск после учета KIM-1, TNFR-1, TNFR-2, MCP-1) в сыворотке крови тесно связаны с ранними структурными изменениями в клубочках, повреждением фенестрированного эндотелия [44]. Увеличение концентрации MCP-1 в моче при ДБП коррелирует с макроальбуминурией и позволяет оценивать прогрессирование поражения почек при СД1 и ожирении [45]. Повышенная экскреция с мочой при ДБП коллагена IV типа отражает повышение синтеза TGF-β, усиление его эффектов и коррелирует с расширением внеклеточного матрикса почек у больных с СД1.
Общедоступным и информативным методом ранней детекции лиц с СД1, подверженных риску ДБП, выступает определение количества в крови лимфоцитов, нейтрофилов, отношения лимфоциты/нейтрофилы [46]. Данные показатели отражают участие лимфоцитов и нейтрофилов в реализации воспалительного процесса в почках при ДБП, коррелируют с отношением альбумина к креатинину в моче.
РОФ как свидетели и участники воспалительного процесса могут быть использованы в качестве маркеров при ДБП у больных СД1. Так, вчС-РБ и сывороточный амилоид А используются в прогнозе прогрессирования диабетической нефропатии, их уровень в сыворотке ассоциирован с микроальбуминурией и связан со смертностью [47]. Концентрация в сыворотке вчС-РБ и AGEs/вчС-РБ у больных с СД1 ассоциирована с изменениями диетического профиля в прогнозе ДБП, сила связи между вчС-РБ в сыворотке и отношением альбумина к креатинину в моче нарастает с увеличением стажа СД [48]. Высокий уровень меди и ЦП в крови является диагностическим маркером и ассоциирован с ухудшением функции почек при СД1 по показателям рСКФ, альбуминурии [37]. ЦП и сывороточный уровень меди – чувствительные маркеры поражения почек при СД1 независимо от альбуминурии [49]. Несмотря на перспективность ЦП в качестве биомаркера за счет участия в воспалении, ОС и метаболизме меди, при ДБП необходимы дополнительные исследования для оценки его чувствительности, специфичности и прогностической ценности в целях диагностики и мониторинга ДБП.
8-оксо-7,8-дигидро-2-дезоксигуанозин – маркер окислительной деструкции ДНК, который выводится с мочой и отражает выраженность ОС наряду с другими показателями редокс-статуса (метилглиоксаль, ТБК-положительные продукты, глутатионпероксидаза, глутатион S-трансфераза, супероксиддисмутаза-1, каталаза, общий антиоксидантный статус), позволяет оценивать прогрессирование ДБП и долгосрочную смертность при СД. Короткий период полураспада активных форм кислорода и азота как прямых маркеров ОС не представляет возможности их детекции при ДБП, определение в сыворотке и/или в моче продуктов окислительной деструкции липидов и белков в качестве биомаркеров прогрессирования диабетической нефропатии является доступным и легким способом оценки выраженности ОС при СД1 [4].
Заключение
Индукция воспаления при сахарном диабете реализуется за счет прямого провоспалительного действия гипергликемии: повреждения клеток нефрона в условиях гиперфильтрации и гемодинамического удара, активации системы комплемента, внутриклеточных сигнальных путей, экспрессии TLR-4, TLR-2, участвующих в синтезе провоспалительных и профибротических медиаторов.
Имеют значение независимые от глюкозы флогогенные механизмы за счет прямых и опосредованных активацией протеинкиназы С эффектов конечных продуктов гликирования, накопления в клетках почек липидов и липотоксичности, окислительного стресса. Окислительный стресс вызывает прямое повреждение клеток и компонентов интерстиция в почечной ткани и активирует воспалительные пути сигнализации (MAPK, NF-κB, JAK-STAT), а также активирует иммунные клетки и способствует синтезу и секреции провоспалительных цитокинов. Реализация воспаления в патогенезе диабетической болезни почек обеспечивается активацией TLR-2 и TLR-4 и формированием NLRP3 инфламмасом, активацией клеток врожденного и адаптивного иммунитета, образованием внеклеточных ловушек нейтрофилов (NET), NET-индуцированным пироптозом клеток почек, активацией системы комплемента по альтернативному и лектин-зависимому пути, синтезом провоспалительных и профиброгенных цитокинов (IL-1β, IL-6, IL-17, IL-18, TNF-α, INF-γ, TGF-β, MCP-1), молекул адгезии (VCAM-1, ICAM-1), реактантов острой фазы (С-реактивный белок, сывороточный амилоид А, церулоплазмин).
Эффекты указанных факторов приводят к расширению мезангиального пространства, утолщению клубочковой базальной мембраны, тубулоинтерстициальному фиброзу, гломеруросклерозу и, как следствие, альбуминурии, протеинурии, прогрессирующему снижению функции почек.
Перспективные биомаркеры воспаления при диабетической болезни почек у больных с сахарным диабетом 1-го типа включают определение концентрации в крови и/или в моче молекулы повреждения почки-1, нейтрофильного желатиназно-ассоциированного липокалина, хемокина MCP-1, TNF-α и его рецепторов TNFR1, TNFR2, количества в крови лимфоцитов, нейтрофилов, отношения лимфоциты/нейтрофилы, концентрации С-РБ, меди и церулоплазмина, 8-оксо-7,8-дигидро-2-дезоксигуанозина, продуктов окислительной деструкции липидов и белков, общего антиоксидантного статуса.
Библиографическая ссылка
Осиков М.В., Эфрос Л.А., Журавлева Л.Ю., Федосов А.А. ВОСПАЛЕНИЕ КАК ФАКТОР ПАТОГЕНЕЗА И ИНСТРУМЕНТ РАННЕЙ ДИАГНОСТИКИ ХРОНИЧЕСКОЙ БОЛЕЗНИ ПОЧЕК У БОЛЬНЫХ С САХАРНЫМ ДИАБЕТОМ ПЕРВОГО ТИПА // Современные проблемы науки и образования. 2025. № 1. ;URL: https://science-education.ru/en/article/view?id=33859 (дата обращения: 08.08.2026).
DOI: https://doi.org/10.17513/spno.33859