


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
GENETIC AND EPIGENETIC ANOMALIES IN SKIN MALIGNANT NEOPLASMS: BASAL CELL AND SQUAMOUS CELL CANCER

Молекулярно-генетическая основа развития злокачественных новообразований кожи включает множество сигнальных путей. На инициацию и прогрессирование опухоли, ведущее к размножению аномальных клеток, влияют генетика, факторы окружающей среды, острые и хронические воздействия, диета, травма и многие другие факторы. Ангиогенез и метастазирование зависят от таких факторов, как возраст и иммунологический статус. Многие факторы окружающей среды, вызывающие злокачественные новообразования кожи, такие как УФ-излучение, сочетаются с генетическими и эпигенетическими изменениями. Новые данные подтверждают роль хронического воспаления в канцерогенезе кожи, опосредованного изменениями в экспрессии и мутациями в генах NF-κB, STAT3 и HIF-1α.
Целью этого обзора является рассмотрение роли генетических изменений во взаимосвязи с 4 биологическими характеристиками рака кожи: аномальной пролиферацией, предотвращением апоптоза, васкуляризацией, неадекватным иммунным ответом. В этом обзоре мы рассмотрим хорошо известные модели, в том числе путь hedgehog для базальноклеточной карциномы и путь p53 (TP53) для плоскоклеточного рака, а также обратимся к новым сигнальным путям, роль которых не полностью выяснена, в том числе для предраковых поражений кожи.
1. Генетика базальноклеточного рака
Немеланомный рак кожи (НМРК, NMSC) включает рак, поражающий кератиноциты, такой как базальноклеточная карцинома (БКК, BCC) и плоскоклеточная карцинома (ПКК, SCC). Этот термин также включает карциному из клеток Меркеля (MCC), редкую нейроэндокринную опухоль и кожную Т-клеточную лимфому (CTCL).
1.1. Факторы и механизмы аномальной пролиферации клеток при БКК
Наиболее распространенным канцерогенным явлением, способствующим НМРК, является ультрафиолетовое излучение (УФ) от воздействия солнца и/или от других источников УФ [1]. Повреждение кожи от воздействия солнца стимулирует онкогенез, повреждая ДНК, создавая воспалительное состояние и активируя реакции окислительного стресса, рецепторные тирозинкиназы (RTK) и антиапоптотические пути [2]. УФ проникает через кожу и повреждает кератиноциты базального эпидермального слоя. UVA проникает глубже в эпителий, воздействуя на строму дермы, тогда как UVB поглощается роговым слоем [3-5]. В первую очередь УФ вызывает повреждение ДНК путем образования димеров циклобутен-пиримидина и 6-4 фотопродуктов [6]. В условиях стресса антиоксидант Sestrin2 активируется опухолевым супрессором р53 и ингибирует mTOR [7-9].
БКК является наиболее распространенным НМРК и составляет 80% всех случаев [10]. Наиболее важным сигнальным путем в онкогенезе БКК является путь Hedgehog (sonic hedgehog (SHH)), состоящий из трех генов SHH, DHH и IHH, а также двух Ptc-генов PTCH1 и PTCH2 [11].
Путь SHH важен для роста и развития позвоночных. Ptc-гены кодируют рецепторы пути SHH, ответственные за подавление Smoothened (SMO), трансмембранного белка/протоонкогена [12]. Лиганд SHH связывается со своим рецептором PTCH (трансмембранным белком), чтобы активировать SMO [13]. Активация SMO вызывает накопление внутриклеточных ионов кальция, что приводит к нарушению кальциевого гомеостаза [14; 15].
После пути SHH точечные мутации гена TP53 являются второй наиболее распространенной генетической мутацией при БКК [11]. Традиционная терапия БКК состояла из хирургической резекции, но появление низкомолекулярных ингибиторов пути SHH позволило использовать новые терапевтические возможности для пациентов с местнораспространенной или метастатической БКК [13; 16].
Активация SHH-сигнального пути на уровне PTCH включает взаимодействие SHH с PTCH через два различных интерфейса: интерфейс между PTCH и кальций/цинксвязывающими поверхностями SHH и интерфейс между PTCH и N-концевым пальмитоилом и C-концевым холестериновым фрагментом SHH. Мутации в этих областях, которые активируют связывание SHH с PTCH, могут стимулировать онкогенез при БКК [17] (рис. 1).
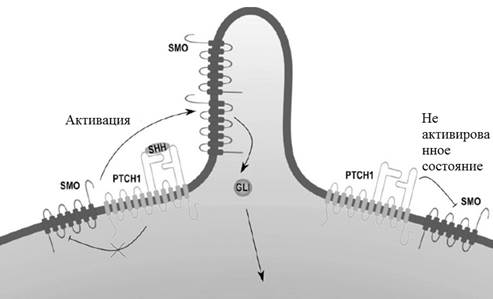
Рис. 1. Сигнальный путь SHH (Sonic hedgehog)
Изменения в системе репарации ДНК. Кератиноциты зависят от функционирования систем репарации ДНК, в частности белков ATM и ATR (члены семейства белков PIKK). После того как клетка подвергается УФ-индуцированному двухцепочечному разрыву ДНК, ATM аутофосфорилируется, что дает ATM возможность фосфорилировать киназу контрольной точки CHK2 [18], которая ингибирует фосфатазу CD25, тем самым препятствуя клеточному митозу. АТМ также может фосфорилировать р53, останавливая клеточный цикл в G1 [19]. Если УФ вызывает разрыв одноцепочечной ДНК, активируется ATR, а затем CHK1 фосфорилирует CDC25 [20]. Кроме того, ATR участвует в p21-пути, который может модулировать различные циклины/CDK для ингибирования клеточного цикла. ATR фосфорилирует MDM2, чтобы инактивировать p53. Инактивация р53 приводит к неконтролируемой пролиферации клеток с поврежденной ДНК. С другой стороны, ATR и ATM могут напрямую фосфорилировать убиквитинлигазу E3 SIAH1, которая активирует p53. Активация p53 посылает клетки, которым в норме должна быть разрешена пролиферация, в апоптоз. Таким образом, дисрегуляция ATR и ATM приводит к непредсказуемой активности клеточного цикла.
Уклонение от апоптоза. Воздействие УФ-излучения активирует стрессовые реакции в эпидермисе, такие как выработка активных форм кислорода, которые повреждают липиды, белки и ДНК, а также активируют антиоксидантную защиту, подавляя онкогенез и инициируя пути апоптоза [21]. Для изучения сигнальных путей, участвующих в окислительном стрессе, рассмотрим далее пути митоген-активируемых протеинкиназ p38 и N-концевых киназ c-Jun (JNK), которые участвуют в про- и антиапоптозных механизмах [22].
Так, p38 представляет собой белок Raf-митоген-активируемой протеинкиназы (MAPK), который может реагировать на окислительный стресс, запуская апоптоз. Когда окислительный стресс активирует систему р38 в кератиноцитах, может активироваться киназа 1, регулирующая сигналы апоптоза. JNK является еще одним членом семейства MAPK, который может быть активирован всего через 5 минут после воздействия УФ-излучения. JNK нацеливается на активирующий белок-1 (AP-1), который является онкогенным фактором транскрипции, участвующим в регуляции клеточного цикла [23]. Фармакологические исследования показали, что ингибирование JNK в кератиноцитах человека in vitro приводит к усилению апоптоза, индуцированного УФ-излучением [24].
Васкуляризация стромы. RTK представляют собой группу рецепторов, активируемых в ответ на воздействие УФ [25]. Некоторые из наиболее известных RTK включают IGF1-R, EGFR, FGFR и VEGFR. RTK и нижестоящие компоненты пути RTK являются мишенями для разработки лекарств [26]. В настоящее время цетуксимаб (ингибитор EGFR) доступен в качестве фармацевтической терапии [27]. Существует два основных пути активации RTK при НМРК: PI3K/mTOR и RAF/MEK/ERK сигнальные пути [26] (рис. 2).
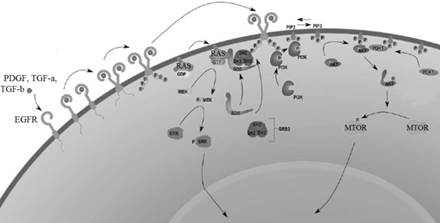
Рис. 2. Сигнальный путь рецепторной тирозинкиназы
Сигнальный путь PI3K/mTOR. Существует две формы mTOR - mTORC1 и mTORC2, и обе участвуют в развитии НМРК [28]. Мутации в генах PI3K и PTEN часто встречаются при НМРК. Чтобы активировать mTORC1, PI3K запускает фосфорилирование субъединицы p85. Это позволяет PI3K фосфорилировать PIP2, превращая его в PIP3. Затем PIP3 рекрутирует фосфоинозитид-зависимую киназу 1 и AKT к рецептору для активации AKT. AKT фосфорилирует негативный регулятор MTORC1, что приводит к активации mTORC1 и в итоге к пролиферации клеток. Активация mTORC2 происходит, когда PI3K фосфорилирует PIP2 до PIP3 [29].
Сигнальный путь FOXO3. Семейство белков FOX «O» представляет собой транскрипционные факторы, которые регулируют продолжительность жизни и подавляют развитие опухолей [30]. FOXO3a транскрипционно нацелен на апоптотические гены, такие как FasL, TRAIL, BIM и PUMA, все из которых участвуют в УФ-индуцированном апоптозе. AKT является негативным регулятором FOKO3a (AKT и его шаперон снижают активность FOXO3a), повышая чувствительность кератиноцитов к апоптозу, индуцированному УФ-В [31].
Сигнальный путь Raf/MEK/ERK. Путь Raf/MEK/ERK также может быть активирован воздействием УФ-излучения. Raf связывает MEK1 и MEK2, фосфорилируя их. MEK1 и MEK2 затем фосфорилируют ERK. ERK1 участвует в регуляции клеточного роста, злокачественной трансформации и лекарственной устойчивости. Исследование показало, что клетки, обработанные ингибиторами SMO, имеют тенденцию к усилению передачи сигналов по RAS/MEK/ERK пути, что приводит к развитию рака [32].
1.2 Аномалии в иммунном ответе. Повреждение кожи, приводящее к воспалительной реакции, также может привести к НМРК. Когда кожа повреждена, микроокружение поврежденного участка изменяется, что позволяет лейкоцитам проникнуть в место повреждения ткани. Сигнальный путь NFκB является хорошо известным участником воспаления. NFκB представляет собой гетеродимер субъединиц p65 и p50, которые связаны с ингибирующим белком IkB. Toll-подобные рецепторы, рецепторы фактора некроза опухоли (TNF) и RTK могут вызывать активацию NFκB путем активации IkB посредством фосфорилирования [33]. Этот процесс устраняет ингибирование NFκB, позволяя белку перемещаться в ядро и способствовать транскрипции провоспалительных генов, таких как TNF-α, IL-1, IL-6, IL-8, и различных других цитокинов и интерферонов. При этом p65-зависимая передача сигналов NFκB поддерживает провоспалительную среду, индуцируя развитие опухоли [33].
Провоспалительные факторы роста и цитокины индуцируют экспрессию фактора транскрипции семейства STAT. STAT3 усиливает транскрипцию факторов, связанных с воспалением, развитием опухоли, выживанием клеток и метастазированием [34]. У мышей, подвергшихся воздействию UVB, сверхэкспрессия STAT3 приводит к ускоренному онкогенезу кожи, тогда как подавление гена STAT3 дает резистентность к образованию опухолей кожи [34].
2. Генетика плоскоклеточного рака
Плоскоклеточный рак кожи (ПРК, cSCC) является одной из наиболее распространенных форм рака кожи во всем мире. cSCC чаще всего выявляют на голове и шее (особенно на лбу, лице, ушах и щеках). Хотя меланома категорически воспринимается обществом как более серьезная угроза, порядка 15 000 пациентов ежегодно умирают от метастатического плоскоклеточного рака в Соединенных Штатах; что превышает общую ежегодную смертность от меланомы [35].
Факторы, вызывающие канцерогенез cSCC, многочисленны и разнообразны; однако основной причиной является воздействие солнечного УФ-излучения. cSCC также может быть инициирован промышленным и неорганическим воздействием сырого парафинового масла, мазута, креозота, смазочного масла, нитрозомочевины и мышьяка в лекарствах, пищевых продуктах и питьевой воде [36]. Кроме того, cSCC может развиваться на фоне хронического воспаления (хронических язв, псориаза, кожной красной волчанки, радиационного дерматита, порокератоза и склероатрофического лихена). Понимание различных генетических путей, в которых возникает cSCC, даст представление об улучшенных и индивидуализированных методах лечения cSCC. Лежащие в основе генетические процессы, участвующие в инициации, стимулировании, поддержании и установлении агрессивных форм роста, напоминают патофизиологические признаки, общие для всех видов рака.
Каждый из канцерогенных путей для cSCC имеет сложные генетические перекрестные взаимодействия между другими сигнальными путями, а также между их компонентами.
УФ-излучение и пролиферация клеток. Сигнальный путь EGFR важен для пролиферации кератиноцитов и заживления ран. В нормальных клетках ETGF-α, TGF-β и PDGF являются 4 из многих лигандов, которые должны связываться с EGFR для его активации. После связывания рецептор подвергается димеризации и аутофосфорилированию, после чего сигнальные белки с доменом SH2 прикрепляются к адаптерному белку Grb2 и комплексу SOS [37]. Этот каскад событий продолжается фосфорилированием пути киназы Ras/Raf/Map, который затем активирует пути mTOR [38]. mTOR вызывает каскад сигнальных событий, кульминацией которых являются комбинации репликации ДНК, синтеза белка и липогенеза. УФ-излучение приводит к мутациям в гене EGFR, способствуя неконтролируемой репликации кератиноцитов и в конечном итоге cSCC. Между BCC и cSCC продемонстрировано сходство по ряду сигнальных путей, модулирующих канцерогенез.
Сигнальный путь NOTCH. Мутации гена NOTCH способствуют развитию cSCC благодаря своей роли в хроническом воспалении. Ген NOTCH отвечает за продукцию белка NOTCH, участвующего в клеточной пролиферации и регуляции апоптоза [39]. Член 1 подсемейства B семейства 1 цитохрома P450 (CYP1B1), гемотиолатмонооксигеназа, участвует в метаболизме и биосинтезе эстрогенов, а также является катализатором гидроксилирования E2 до 4-гидроксиэстрадиола. Было обнаружено, что нормальные кератиноциты экспрессируют этот фермент. Фактически, недавние исследования показали, что CYP1B1 противодействует передаче сигналов NOTCH1, который, в свою очередь, блокирует пролиферацию и дифференцировку кератиноцитов. В настоящее время проводятся дополнительные исследования влияния CYP1B1 и NOTCH1 на клетки SCC. RBPJ ещё один ген сигнального пути NOTCH. Этот ген кодирует фактор транскрипции RBP-Jκ, также известный как «CBF1, Suppressor of Hairless, Lag-1» (CSL) в кератиноцитах. Верхние эпителиальные клетки подавляют продукцию CSL. Однако предзлокачественные и cSCC демонстрируют повышенный уровень CSL, что приводит к избыточной пролиферации нормальных эпителиальных клеток, лежащих в основе развития актинических кератозов и cSCC [40].
Роль микроРНК. МикроРНК (миРНК) играют важную роль в пролиферации клеток cSCC. Однако функция многих микроРНК варьирует. В различных сигнальных путях одни микроРНК активируются, тогда как другие подавляются, вызывая различные клеточные ответы. Например, miR-21 действует как онкоген, нацеленный на фактор транскрипции GRHL3, важный элемент сигнального пути PTEN. Когда miR-21 активируется, компоненты сигнального пути PTEN и PI3K/Akt/mTOR увеличивают свою экспрессию, что приводит к усилению клеточного роста [41]. Это одна из многих микроРНК, которые влияют на рост клеток cSCC. Некоторые из микроРНК с повышенной экспрессией определяют агрессивность поражения cSCC. Поскольку микроРНК влияют как на путь PTEN, так и на путь mTOR, всегда остается возможность обнаружить схожие изменения между сигнальными путями БКК и ПРК.
Уклонение от апоптоза. Как УФ-А, так и УФ-В излучение вызывают наиболее распространенные типы cSCC. Хорошо известно, что УФ-В-излучение вызывает прямое повреждение ДНК кератиноцитов, приводящее к cSCC, но также было показано, что воздействие УФ-А-излучения способствует онкогенезу за счет повреждения ДНК из-за присутствия фотосенсибилизаторов в организме, вызывающих косвенное производство АФК. Солнечное УФ-излучение может вызывать мутации в ДНК в димерах циклобутен-пиримидина [42]. Эти мутации могут активировать систему репарации ДНК ATR, которая, в свою очередь, активирует TP53, опухолевой супрессор, важный для апоптоза. Мутация в одном или обоих генах может привести к ускользанию от апоптоза и, как следствие, к сверхактивному росту клеток. Мутации TP53 вызывают неконтролируемую активацию клеточного цикла, позволяя мутировавшим клеткам преждевременно переходить в фазу синтеза (фаза S) клеточного цикла. CDKN2A/p16 дикого типа отвечает за образование ингибиторов циклинзависимой киназы (CDK) INK4A и p14, оба из которых являются ингибиторами деградации TP53. INK4A связывается с cdk4 и cdk6, ингибируя переход клеточного цикла в S-фазу. Когда онкоген CDKN2A/p16 мутирует, мутирует INK4A, что приводит к отсутствию ингибирования cdk4 и cdk6 в клеточном цикле. Этот механизм напоминает механизм TP53 в том, что клеточный цикл преждевременно переходит в фазу S. (рис. 3).
Снижение экспрессии некоторых микроРНК может влиять на апоптоз; в частности, подавление miR-34a снижает активность TP53, что приводит к предотвращению апоптоза. Отдельные кератиноциты, облученные солнечным УФ-излучением, по-видимому, имеют подавленную экспрессию miR-34a, что приводит к проапоптотическому эффекту в этих опухолевых клетках [41].
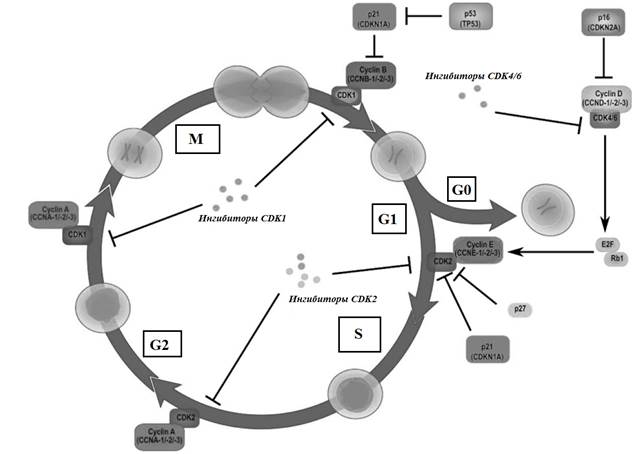
Рис. 3. Клеточный цикл, демонстрирующий TP53-ингибирование клеточной пролиферации
Предраковые поражения кожи и уклонение от иммунного ответа. Кератоакантомы (КА) являются предраковыми поражениями. Эти поражения классифицируются как поражения с потенциалом cSCC, которые могут спонтанно разрешаться. Тем не менее ожидание спонтанного разрешения КА может быть вредным, поскольку не все поражения регрессируют. Некоторые КА продолжают быстро расти; могут стать агрессивными и метастазировать. Существует множество генных мутаций, вызывающих трансформацию сигнальных путей, которые в конечном итоге приводят к появлению этих поражений. Наиболее распространенным типом мутирующего генетического пути является путь апоптоза. Наиболее примечательными генами с повышенной экспрессией, обнаруженными с помощью микрочипового анализа в KA, были MALAT-1, S100A8 и EHF. Эти гены модулируют каспазы, Bax и Bcl-2, что приводит к предотвращению апоптоза [43]. Микрочипы показывают более 1000 генов, которые отличаются от таковых для cSCC, однако между cSCC и KA есть много сходных гистологических особенностей.
Излучения UV-B и UV-A могут оказывать влияние на иммунный ответ. Актинический кератоз (АК) представляет собой предраковое поражение, которое может развиться в плоскоклеточный рак. Следует ли рассматривать АК как диспластическое, предраковое поражение кератиноцитов или их следует понимать как сильно рассеянный, дезорганизованный cSCC in situ, оспариваемый факт в сферах дерматопатологии и гистопатологии. Для выяснения правильной классификации АК потребуются дальнейшие исследования. В данном обзоре АК будут считаться предраковыми поражениями. Прогрессирование от AK до cSCC связано с модуляцией сигнальных путей иммунной системы UV-A и UV-B излучениями. УФ-излучение вызывает усиление хронического воспаления, связанного с p53, влияющего на апоптоз кератиноцитов; однако только примерно 10% AK прогрессируют до cSCC. Согласно одной из концепций, судьба АК зависит от пошагового прохождения нескольких стадий, обусловленных сложными иммунологическими механизмами. Во многих случаях АК могут протекать бессимптомно с небольшим воспалением или без него. Эти поражения можно подразделить на бессимптомные (асимптомные) актинические кератозы (ААК). Считается, что некоторые ААК прогрессируют по мере того, как клетки претерпевают молекулярные и иммунологические изменения, которые способствуют усилению роста и воспалению. Например, десенсибилизация сайта Fas-лиганда (FasL) может приводить к ингибированию CD8 + Т-лимфоцитов, приводя к нарушению CD8 + -опосредованного апоптоза и, таким образом, способствовать прогрессированию поражения [41]. Другие ААК могут не претерпевать изменений, необходимых для прогрессирования, и оставаться бессимптомными.
УФ-индуцированное повреждение кожи вызывает каскад заживления ран, активируя пролиферацию кератиноцитов и многие пути клеточной передачи сигналов, которые участвуют в онкогенезе. Путь EGFR, внутренний и внешний пути воспаления включены в клеточные сигнальные пути, вызывающие онкогенез. Хронические воспалительные состояния, в том числе склероатрофический лихен, могут активировать внутренние пути, вызывающие хроническое воспаление, повышенный уровень обновления клеток и возможный онкогенез [44]. Внешний путь включает влияние факторов окружающей среды и вирусных причин воспаления, включая вирус папилломы человека (ВПЧ) [44] (рис. 4). Этот путь включает высвобождение каскада воспалительных цитокинов и хемокинов, которые способствуют миграции лимфоцитов к месту повреждения и последующему восстановлению. Хемокины могут усиливать клеточное обновление, вызывая повышенную репликацию ДНК, предрасполагая клетки к генетическим мутациям, которые способствуют образованию опухолей, таких как cSCC [44].
Кожный односторонний порокератоз, еще одно хроническое воспалительное заболевание, обладает высоким потенциалом злокачественной трансформации из-за мутаций в псориазине, p16 и инволюкрине, сходными с таковым при cSCC, но на более низком уровне [45]. Однако, когда поражения AK и cSCC действительно появляются, они продуцируют хемокины, которые увеличивают метастатический потенциал.
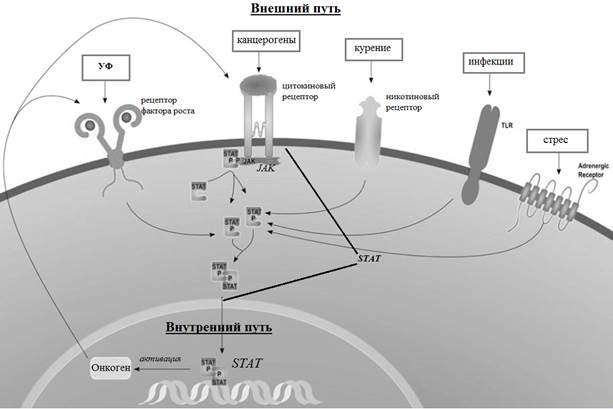
Рис. 4. Показаны повреждающие факторы внешнего пути (инфекция, стресс, УФ-излучение и канцерогены) и внутренний путь, обусловленный активацией онкогена
Химически индуцированный мутагенез при ПКК (cSCC). Химические вещества, в том числе мышьяк и фармакологические агенты, могут быть вовлечены в развитие cSCC. Хроническое воздействие высоких концентраций мышьяка в пищевых продуктах и питьевой воде является частой причиной ПКР, особенно ПКР на ладонях. Было обнаружено, что уровни мышьяка в питьевой воде всего 300 мкг/л вызывают cSCC [46]. Есть много лекарств и прививок, которые включают использование мышьяка в качестве адъювантов, активных и неактивных ингредиентов в зависимости от лекарства или вакцины; однако количества этих фармакологических агентов недостаточно, чтобы вызвать злокачественную трансформацию кератиноцитов. Тем не менее необходимы дальнейшие исследования хронического воздействия небольших количеств мышьяка и возможных злокачественных новообразований. Экспонированные мышьяком гиперкератотические эпителиальные клетки экспрессируют повышенные уровни кератина-1, кератина-10, инволюкрина и лорикрина, биохимических медиаторов, важных для пролиферации кератиноцитов [46]. Эти поражения могут в дальнейшем прогрессировать, превращаясь в болезнь Боуэна, а затем в cSCC. Также было обнаружено, что мышьяк изменяет функциональность факторов транскрипции и коактиваторов транскрипции, которые влияют на рост клеток и стромальную среду, в которой находятся кератиноциты. УФ-индуцированные cSCC обычно экспрессируют мутантные TP53, HRAS или другие мутантные гены-супрессоры опухоли, приводящие к уклонению от апоптоза. Однако при анализе модуляции сигнальных путей, вызванной мышьяком, выявлены другие локусы (Nrf-2, Yap и др.) [46].
Ингибиторы BRAF также могут влиять на появление cSCC. Во время лечения ингибиторами BRAF могут спонтанно появляться cSCC. Есть много сообщений о случаях, показывающих, что ингибитор BRAF, сорафениб, ингибирует PI3K, MAP-киназу и NFκB, что может снижать высвобождение цитокинов из клеток Лангерганса, уменьшая ответ Т-лимфоцитов на новые cSCC [47].
Мышьяк и УФ-излучение одинаково воздействуют на строму кератиноцитов. Хроническое воздействие мышьяка приводит к гиперсекреции ММП, которые разрушают коллаген IV типа в базальной мембране. Это способствует диапедезу иммунных клеток и высвобождению воспалительных маркеров и хемокинов, вызывая дальнейший рост клеток и повышенный злокачественный потенциал. В дополнение к мышьяку УФ также влияет на строму нижележащих кератиноцитов. УФ-излучение приводит к генетическим изменениям внутри клетки, затрагивающим дермальные фибробласты и нижележащие базальные мембраны. Структура микроокружения, включая базальную мембрану эпидермиса и внеклеточный матрикс дермы, может быть изменена предраковыми поражениями. Эти предраковые поражения вызывают высвобождение хемокинов, что приводит к инвазии воспалительных клеток и стимулирует рост cSCC. Было обнаружено, что AK и cSCC имеют повышенное содержание MMP, дезинтегриноподобного домена металлопротеиназ (ADAM), тканевых ингибиторов металлопротеиназ и других ферментов, разрушающих внеклеточный матрикс. Кроме того, опухолевые клетки изменяют молекулярную функцию межклеточной адгезии и предотвращают связывание интегринов поврежденной базальной мембраны с внеклеточным матриксом и связывание с опухолевыми клетками. Опухолевые клетки усиливают активность фактора комплемента Н и фактора Н-подобного белка 1, что приводит к дальнейшему поражению и инвазии. Активация этих воспалительных сигналов опухолевыми клетками напрямую влияет на микроокружение кератиноцитов, вызывая дальнейшее повреждение стромы, что приводит к инвазии воспалительных клеток и стимулирует рост cSCC [48].
Травмы и хронические воспалительные состояния кожи. Хроническое воспаление и травма могут изменить эпигенетику нижележащей стромы (на которой лежат эпителиальные клетки), позволяя этим клеткам иметь больший метастатический потенциал. Активация STAT3, p63, FGFR2 и других генов, кодирующих и активирующих хемокины, может быть изменением, необходимым для приобретения инвазивного потенциала [48]. Хемокин SDF-1 и его эндотелиальный клеточный рецептор хемокинового рецептора CXC типа 4 (CXCR4) вовлечены в ангиогенез и активируются при хронических воспалительных состояниях. Как только формируются предраковые поражения, SDF-1 и CXCR4 могут влиять на хемотаксис, повышающую регуляцию металлопротеинов и активацию стромальных фибробластов, что приводит к потере коллагена в базальной мембране [47; 48]. Язвы Маржолина (наблюдаемые у ожоговых больных), хронические пролежни и диабетические язвы могут подвергаться процессам, которые приводят к стромальным изменениям и последующей дифференцировке кератиноцитов. Сигнальные пути, связанные с трансформацией хронической воспалительной язвы, могут возникать из спонтанно нового пути, который также связан с ростом эпителиальных клеток. Один из таких сигнальных путей связан с ингибированием cdk, PDGF и SHH. Другой связан с подавлением проапоптотического пути WNT/β catenin. Оба пути подавляют гены внеклеточного матрикса и активируют активацию генов MMP, влияя на многие из тех же путей, на которые влияет УФ-излучение.
Не всем cSCC предшествуют предраковые поражения. Некоторые cSCC могут появиться de novo. Это особенно верно в отношении cSCC, развившихся у пациентов, перенесших трансплантацию. Пациенты с трансплантацией часто имеют ослабленную иммунную систему как из-за иммуносупрессивной терапии, так и из-за основных состояний, вызвавших необходимость трансплантации. Было обнаружено, что у пациентов с ПКР после трансплантации мутации Т-лимфоцитов de novo в ZNF577, кодирующем белки цинковых пальцев, и гене FLOT, кодирующем миграцию Т-клеток, ослабляют иммунный ответ против опухолевых клеток и последующий избыточный рост. Эти генетические мутации, которые приводят к cSCC, должны быть исследованы для разработки инновационных подходов к лечению пациентов с продвинутыми формами cSCC [49].
Таким образом, онкогенез cSCC сложен. Воздействие УФ-излучения, воздействие канцерогенных веществ, изменение стромального окружения кератиноцитов и хронические воспалительные состояния могут быть вовлечены в развитие опухоли независимо или в сочетании. Существует несколько возможных генетических драйверов, способствующих развитию cSCC. Каждый из этих факторов может влиять на развитие злокачественного новообразования, воздействуя на один из четырех характерных патофизиологических процессов, общих для всех видов рака. В зависимости от провоцирующих факторов, мутаций в сигнальных путях cSCC может иметь различную степень агрессивности и требовать различных видов терапии. Важно отметить, что все опухолевые клетки не одинаковы. На самом деле мутации и изменения в сигнальных путях, рассмотренные выше, могут происходить одновременно внутри одной опухоли в нескольких клетках.
Заключение
Знание молекулярных сигнальных путей немеланомного рака кожи может помочь исследователям и медицинским сотрудникам предоставить больше терапевтических возможностей пациентам, страдающим от этих заболеваний. Обладая этой информацией, можно блокировать провоцирующие генетические факторы с помощью фармако- и иммунотерапии, что позволяет лечить пациентов с терапевтически резистентными, местнораспространенными и даже метастатическими поражениями за счет различных вариантов терапии, блокирующих множественные сигнальные пути. Хотя известна некоторая информация о генетических путях развития злокачественных новообразований кожи, по-прежнему существует потребность в исследованиях, которые можно провести для улучшения вариантов лечения, чтобы снизить уровень смертности и рецидивов.
Существуют различные провоцирующие факторы, вызывающие кожные новообразования. Совокупное воздействие всех факторов (экспосома) изменяет экспрессию генов, что приводит к неконтролируемому росту. В случае кожных злокачественных новообразований солнечное УФ-излучение рассматривается как канцероген, который может действовать либо как стимулятор (инициатор), либо как активатор, в зависимости от степени воздействия и защитных мер.
МикроРНК играют важную роль в сигнальных путях, определяющих активацию и супрессию специфических генов транскрипционных факторов. МикроРНК могут вызывать гипометилирование генов, что приводит к усилению транскрипции, трансляции и неконтролируемой пролиферации. Многие из этих генов активируются и подавляются, оказывая различное влияние на клетки; эти эффекты могут также привести к появлению и прогрессированию рака кожи. Существуют сотни и тысячи микроРНК, которые могут трансформировать функцию генетических путей и факторов транскрипции с нормально функционирующего пути на злокачественный путь. Эта информация может помочь при дальнейшем тестировании пациентов и, возможно, в качестве профилактики для предотвращения появления кожного злокачественного новообразования.
Опухолевые клетки, как и иммунные клетки, могут обладать способностью высвобождать хемокины, что приводит к аутокринной и паракринной связи между клетками. Этот процесс также может формировать систему связи между клетками, сообщая каждой из них их функцию, и даже проникать в доброкачественные клетки, делая их злокачественными. Эти данные дают медицинским сотрудникам дополнительную информацию для проведения обследований кожи и наблюдения за хроническими воспалительными состояниями пациентов на предмет трансформации в злокачественные опухоли. А также являются основой для разработки персонализированного динамического молекулярного тестирования для выявления критического набора измененных онкогенетических путей.
Библиографическая ссылка
Сустретов В.А., Кутилин Д.С., Максимов А.Ю., Шатова Ю.С., Касьяненко В.Н. ГЕНЕТИЧЕСКИЕ И ЭПИГЕНЕТИЧЕСКИЕ АНОМАЛИИ ПРИ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЯХ КОЖИ: БАЗАЛЬНОКЛЕТОЧНЫЙ И ПЛОСКОКЛЕТОЧНЫЙ РАК. // Современные проблемы науки и образования. 2022. № 4. ;URL: https://science-education.ru/en/article/view?id=31793 (дата обращения: 27.07.2026).
DOI: https://doi.org/10.17513/spno.31793