


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
THE HEREDITARY PROGRESSIVE LIMB-GIRDLE MUSCULAR DYSTROPHY TYPE 2L (ANOCTOMINOPATHY)

Поясно-конечностные мышечные дистрофии (ПКМД) – генетически гетерогенная группа заболеваний с выраженным клиническим полиморфизмом, характеризующаяся прогрессирующей слабостью мышц преимущественно тазового и плечевого поясов.
ПКМД имеют аутосомно-доминантный (ПКМД 1 типа) и аутосомно-рецессивный типы наследования (ПКМД 2 типа). Мутации в одном и том же гене могут вызывать разные фенотипические варианты миопатий. К наиболее частым ПКМД в зависимости от мутации в определенных генах относятся кальпаинопатии, дисферлинопатии, саркогликанопатии, ламинопатии, аноктаминопатии и т.д. [1]. Аноктамин 5 (ANO5) является внутриклеточным кальций-зависимым хлоридным каналом, который экспрессируется в скелетных мышцах, миокарде и костях. Его функция до конца не ясна, но известно, что он локализуется в эндоплазматическом ретикулуме и может принимать участие в репарации мышечного волокна после повреждения [2]. Рецессивные мутации в гене ANO5 приводят к таким клиническим фенотипам первичных скелетно-мышечных заболеваний как ПКМД 2L и дистальная Миоши-подобная миопатия, которые впервые были описаны в 2010 году в 5 семьях [3, 4]. Фенотипически они имеют сходства с дисферлинопатиями, которые вызваны мутацией в гене дисферлина (DYSF) и включают в себя два основных фенотипа – ПКМД 2B и дистальную миопатию Миоши, а также различные варианты от бессимптомной гиперКФКемии до более тяжелых форм [3]. Аутосомно-доминантная мутация в гене ANO5 приводит к поражению костей – гнатодиафизарная дисплазия [5]. Мутации c.191dupA в 5 экзоне и c.2272C>T в 20 экзоне гена ANO5 являются наиболее частыми среди всех мутаций при ПКМД 2L в европейской популяции [6]. В настоящее время выявлены различные мутации в экзонах 5, 8, 13, 14, 16, 18 и 20 данного гена. ПМКД 2L встречается в 2 раза чаще ПКМД 2В и занимает третье место по распространённости после ПКМД 2A и ПКМД 2I среди европейского населения, как и саркогликанопатии [7, 8]. В азиатской популяции ПМКД 2L встречается очень редко, о чем свидетельствуют данные когортного исследования, проведенного в Китае [8]. В зарубежной литературе приводятся описания как единичных клинических случаев, так и групп пациентов с ПКМД 2L. У большинства заболевание начинается со слабости проксимальных мышц нижних конечностей, гипотрофии бедер и голеней, а первые симптомы связаны с трудностями при ходьбе и подъеме по лестнице. У всех пациентов слабость в нижних конечностях более выражена, чем в верхних, многие из них до появления симптомов активно занимались спортом. Возраст начала заболевания варьирует от 20 до 55 лет. Женщины имеют более доброкачественное течение болезни. Уровень КФК в крови превышает норму в 10-40 раз, но в единичных случаях лишь незначительно повышен [4,5]. Помимо классической формы с вовлечением проксимальных мышц конечностей, существуют и другие фенотипы: дистальная миопатия (по типу Миоши) с атрофией икроножных мышц, изолированной проксимальной мышечной слабостью нижних конечностей; изолированная проксимальная мышечная слабость в верхних конечностях; проксимально-дистальная мышечная слабость в нижних конечностях; изолированная гипертрофия икроножных мышц; асимптомная КФКемия [5]. Часто наблюдается сниженная толерантность к физическим нагрузкам и миалгии [9]. Четверть пациентов теряют способность самостоятельно передвигаться и нуждается в инвалидном кресле спустя 10-20 лет от начала болезни [1,10]. K. Wahbi и соавт. в 2013 году впервые описали вовлечение сердечной мышцы в патологический процесс при этом заболевании, о чем ранее не было известно. Они показали, что дилатационная кардиомиопатия может быть проявлением мутации в гене ANO5. Однако количество пациентов не позволило провести реальную оценку распространенности кардиомиопатии при ПКМД 2L. Все же следует быть настороженным при выявлении у пациента повышенного КФК в сыворотке крови и кардиомиопатии, не связанной с такими заболеваниями, как гипертоническая болезнь, ишемическая болезнь сердца и т.д. [11] При ПКМД 2L имеются относительно характерные паттерны мышечной визуализации. Наблюдается гипотрофия мышц задних отделов бедер и икр с замещением их жировой тканью, менее выраженная гипотрофия мышц передних отделов бедер; прямая, портняжная, тонкая мышцы бедра и короткая головка двуглавой мышцы бедра часто остаются сохранными и вовлекаются в патологический процесс на более поздних стадиях [5, 6, 12]. При биопсии мышц характерны неспецифические дистрофические изменения, кроме того, при окраске Конго красным выявляются отложения амилоида в мышечной стенке кровеносных сосудов и в интерстиции, раннее это было описано при дисферлинопатиях, причем у данных пациентов не было признаков системного амилоидоза [10]. В когортном исследовании, в которое включили 101-го пациента с неуточненной миопатией, у 25 была выявлена мутация в гене ANO5 [6]. Учитывая это, можно сделать вывод, что при миопатиях неуточненного генеза следует проводить дифференциальный диагноз с аноктаминопатиями.
Цель исследования: описание клинического случая генетически подтвержденной ПКМД 2L типа с поздним началом.
Клиническое наблюдение
Пациентка М., 55 лет поступила в январе 2020 года в неврологическое отделение Национального Медицинского Исследовательского Центра (НМИЦ) им. В.А. Алмазова с жалобами на нарушение походки, трудности при вставании с низкого стула, подъеме по лестнице, снижение толерантности к физическим нагрузкам, боль в мышцах бедер. В анамнезе пациентки обращала на себя внимание отягощенная наследственность (боковой амиотрофический склероз по материнской линии). Кроме того, пациентка страдала ожирением I степени и гипертонической болезнью II стадии. В молодом возрасте пациентка занималась конькобежным спортом и никаких трудностей во время занятий спортом не отмечала. Первые симптомы появились в начале 2013 года в возрасте 48 лет, когда знакомые пациентки стали обращать внимание на изменение ее походки. Чуть позже сама пациентка заметила появление трудности при подъеме по лестнице, постепенно появилась и стала нарастать неловкость в левой ноге. В сентябре того же года после вакцинации от гриппа отметила появление субфебрилитета в течение месяца. Была обследована в отделении неврологии по месту жительства, где было выявлено повышение уровня креатинкиназы (КФК) до 1509 ЕД/л (N 24,00-170,00), по данным электронейромиографии (ЭНМГ) были установлены выраженный текущий миогенный процесс и полинейропатия смешанного генеза с поражением проксимальных отделов нижних конечностей. Была назначена терапия (Октолипен, Нейромидин, витамины группы В), однако эффекта от лечения отмечено не было. Постепенно до января 2014 года слабость в ногах нарастала, несколько асимметрично, больше слева, пациентка стала отмечать трудности при вставании из положения сидя, для чего ей требовалась помощь рук. В январе 2014 года пациентка была госпитализирована в отделение ревматологии по месту жительства, где на основании уже имевшихся данных и проведенного дообследования (повышение КФК до 1509 ЕД/л, уровень циркулирующих иммунных комплексов (ЦИК) до 103 у.е. (N 0-120)) был установлен диагноз: первичный идиопатический полимиозит, подострое течение, активность 2, полинейропатия смешанного генеза с преимущественным вовлечением нижних конечностей. Была проведена пульс-терапия глюкокортикостероидами (ГКС) – метилпреднизолоном (750 мг), с некоторым положительным эффектом. В марте того же года в ревматологическом отделении другого стационара был выставлен иной диагноз: системный васкулит, и проведено лечение преднизолоном в дозе 60 мг в сутки. На фоне монотерапии ГКС слабость в ногах сохранялась и значимой динамики не отмечалось. Пациентка была снова госпитализирована в отделение ревматологии по месту жительства в июне 2015 года. При обследовании установлено отсутствие антинуклеарного фактора (АНФ) и антител к цитоплазме нейтрофилов, однако отмечено нарастание КФК до 2500 ЕД/л. С представлением об узелковом периартериите, смешанной полинейропатии с миопатическим синдромом была назначена терапия ГКС (метилпреднизолон по 250 мг №4) и цитостатиками (ЦС) (циклофосфан 400 мг №3), на фоне которой пациентка впервые отметила уменьшение слабости в ногах. Было рекомендовано продолжение данной терапии амбулаторно с постепенным снижением дозы, однако пациентка после выписки вновь отметила ухудшение состояния и рекомендации не соблюдала. Тем не менее, при повторной госпитализации в августе 2016 года лабораторные данные свидетельствовали о снижении уровня КФК до 1494 ЕД/л, при этом установлено повышение миоглобина 382 нг/мл (N 0,0-106,0) и лактатдегидрогеназы (ЛДГ) 627 ЕД/л (N 125-220). Была проведена биопсия кожно-мышечного лоскута: достоверных признаков миозита, васкулита не выявлено, однако нельзя было исключить миопатическую дистрофию. Был выставлен диагноз: миопатический синдром неуточненного генеза, сенсо-моторная полинейропатия с преимущественным вовлечением нижних конечностей, и проведен очередной курс терапии ГКС (преднизолон 5 мг в сутки с продолжением на амбулаторном этапе) и цитостатиками (эндоксан 2500 мг на курс) без значительной положительной динамики. Для коррекции терапии в 2017 году года пациентка поступила в ревматологическе отделение НМИЦ имени В.А. Алмазова. С представлением о полимиозите умеренной степени активности, и учитывая эффективность использования ЦС в анамнезе, было принято решение о возобновление терапии циклофосфаном (до достижения суммарной дозы 3000 мг) совместно с ГКС (преднизолон 5 мг). Терапию пациентка переносила удовлетворительно, однако клинически и лабораторно значимого эффекта достигнуто не было. Была произведена смена циклофосфана на циклоспорин (от 50 до 150 мг в сутки), которые затем были заменены метотрексатом (20 мг 1 раз в неделю), также без положительного клинического эффекта. Пациентка стала отмечала боли в мышцах бедер, быструю утомляемость в мышцах ног, нарушение походки. Лабораторно также сохранялись лабораторные признаки активности заболевания (миоглобин 615 нг/мл, КФК 1970 Ед/л, ЛДГ 303 Ед/л). С учетом рефрактерного течения полимиозита в апреле 2018 года было принято решение инициировать анти-В-клеточную терапию ритуксимабом. На первую и вторую инфузии по 500 мг была отмечена хорошая переносимость и положительная динамика клинических и лабораторных данных, поэтому было рекомендовано продолжить ритуксимаб в дозировке 1000 мг раз в полгода с сохранением прежних доз ЦС и ГКС. Однако в декабре 2018 года было вновь установлено нарастание активности симптомов, в связи с чем была увеличена дозировка ритуксимаба до 2000 мг, но в дальнейшем лабораторные данные от мая 2019 года (КФК – 1722 Ед/л, миоглобин – 326,5 нг/мл, ЛДГ – 281 Ед/л) свидетельствовали о неэффективности проводимой медикаментозной терапии ритуксимабом. Была выполнена магнитно-резонансная томография (МРТ) мышц бедра, выявившая картину выраженной атрофии и жировой дистрофии всех групп мышц бедер (больше правого бедра), умеренного миозита задней и медиальной групп мышц бедер. Для верификации поражения мышц проводилась биопсия мышечного лоскута с передней-боковой поверхности левого бедра: отмечена инвазия лимфоцитов и нейтрофильных гранулоцитов в цитоплазму отдельных мышечных волокон с явлениями фокального некроза и признаки хронического поражения мышц в виде фиброза и липоматоза. Была проведена постепенная отмена ЦС и ГКС, однако после полного прекращения терапии пациентка отметила некоторое ухудшение состояния.
Для дообследования и подбора терапии пациентка поступила в отделение неврологии НМИЦ с вышеописанными жалобами. В неврологическом статусе при поступлении был выявлен псевдобульбарный синдром (хоботковый, Маринеску-Радовичи с двух сторон); мышечный тонус был диффузно понижен, больше в нижних конечностях, пальпация мышц безболезненна. При исследовании мышечной силы отмечалась слабость всех мышц бедер с обеих сторон до 3-4 баллов, более выраженная слева, легкая уступчивость грудных мышц и мышц-разгибателей пальцев рук с двух сторон. Отмечались гипотрофии мышц бедер, кистей рук и псевдогипертрофии мышц голеней с 2-х сторон. Фасцикуляций не наблюдалось, однако на фоне нагрузки в мышцах бедер появлялись крампи. Брюшные рефлексы были сохранны; сухожильные рефлексы с рук были симметрично снижены; поверхностные и глубокие рефлексы с нижних конечностей отсутствовали. Наблюдались патологические стопные знаки Пусепа с двух сторон. Кроме гипестезии в области тыльной и ладонной поверхности 4-го и 5-го пальцев кистей с двух сторон, других нарушения чувствительности пациентка не предъявляла. При проведении координаторных проб отмечалась легкая интенция в нижних конечностях. В позе Ромберга определялась шаткость, вероятно, за счет парезов. При оценке осанки и походки у пациентки были отмечены характерная поза с переразгибанием бедер и усилением поясничного лордоза (рис. 1), миопатический паттерн ходьбы.
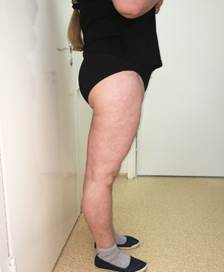
Рис. 1. Нарушения осанки и характерная поза с переразгибанием бедер, усилением поясничного лордоза
При проведении лабораторных исследований было выявлено повышение КФК до 1618 ЕД/л и обнаружен АНФ титром 1:100 с гранулярным типом свечения. При проведении ЭНМГ верхних и нижних конечностей была подтверждена компрессионно-ишемическая невропатия локтевых нервов с двух сторон и изменения миогенного характера во всех исследованных мышцах, наиболее выраженные в мышцах бедер. По данным МРТ мышц прямая, портняжная, тонкая мышцы были относительно сохранны с незначительным отеком, полусухожильная мышцы слева была в состоянии выраженного отека со скоплением жидкости под фасцией мышцы. Остальные мышцы фактически полностью заменены жировой тканью при сохранении объема передней группы и с признаками гипотрофии задней группы (рис. 2).
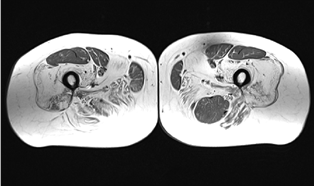
Рис. 2. МРТ-картина выраженной дегенерации мышц бедер с замещением жировой тканью
С учетом миопатического синдрома были проведены спирография и эхокардиография, по результатам которых деятельность дыхательных мышц и сердца оказались не нарушены. Учитывая медленно прогрессирующее течение заболевания с преимущественным поражением мышц тазового пояса и проксимальных отделов ног, стабильно высокий уровень КФК в период всего заболевания (от 1500 до 2000 Ед/л), отсутствие клинического и лабораторного эффекта от проводимой терапии ГКС, ЦС и анти-В-клеточной терапии, данные биопсии мышечного лоскута с отсутствием достоверных признаков воспалительного процесса, а также выраженные признаки хронического поражения мышц был предположен диагноз наследственного заболевания - мышечной дистрофии. По результатам исследования активность измеренных лизосомных ферментов была в пределах референсных значений, что позволило исключить гликогеноз II типа. Методом массового параллельного секвенирования на приборе Ion S5 был проведен анализ 82 генов, ответственных за метаболические и поясно-конечностные мышечные дистрофии. Были выявлены изменения нуклеотидной последовательности: в 5 экзоне гена ANO5 выявлена мутация c.242A>G (p.Asp81Gly) в гетерозиготном состоянии, а в 20 экзоне гена ANO5 – мутация c.2272C>T (p.Arg758Cys), также в гетерозиготном состоянии. В соответствии с критериями ACMG (American College of Medical Genetics) выявленные варианты нуклеотидной последовательности являются патогенными вариантами. Обе мутации описаны при фенотипе ПКМД 2L, мутация в гене ANO5 ассоциирована также с дистальной миопатией Миоши. Таким образом, пациентке был установлен основной диагноз: поясно-конечностная мышечная дистрофия 2L типа. Проксимальный нижний умеренный парапарез. С учетом наследственного характера и медленного прогрессирования данного заболевания, пациентке показана неспецифическая поддерживающая терапия, лечебная гимнастика, массаж и ортопедические мероприятия, а также динамическое наблюдение, особенно с учетом склонности миодистрофий к поражению дыхательной и сердечной мускулатуры.
Заключение
В представленном нами случае ПКМД 2L типа манифестировала у пациентки в достаточно зрелом возрасте (48 лет). От появления первых симптомов до постановки диагноза прошло 7 лет. Длительный период дифференциальной диагностики сопровождался ошибочными представлениями о наличии аутоиммунного заболевания с поражением мышц (полимиозита), что было сопряжено с назначением иммуносупрессивной терапии, ответ на которую был минимален и в конечном итоге отсутствовал. По данным литературы существует гипердиагностика полимиозита, и при отсутствии должного ответа на иммуносупрессивную терапию следует пересмотреть представление о диагнозе. В вышеописанном наблюдении против воспалительной миопатии также свидетельствовали данные мышечной биопсии, где воспалительные изменения мышц были представлены минимально. Диагноз мышечной дистрофии подтверждали данные МРТ мышц, выявившие признаки жировой дегенерации мышц бедер. Укладывается в представление о наследственной миодистрофии и уровень КФК у нашей пациентки, который превышал норму до 10 раз, но на всем протяжении заболевания был на достаточно стабильном уровне. Отсутствие отложения амилоида в мышцах при окраске Конго красным, характерное для ПКМД 2L [10], не могло в данном случае повлиять на достоверность диагноза, так как решающую роль в постановке диагноза ПКМД 2L сыграли результаты молекулярно-генетической диагностики. Функция сердечной мышцы у представленной пациентки нарушена не была, однако следует проводить периодически скрининговое исследование, учитывая возможное развитие дилатационной кардиомиопатии у пациентов с ПКМД 2L [10].
Таким образом при миопатии неуточненного генеза с поздним началом с преимущественным поражением проксимальных мышц конечностей следует быть настороженными в отношении ПКМД и проводить молекулярно-генетическое исследование.
Библиографическая ссылка
Исабекова П.Ш., Алексеева Т.М. НАСЛЕДСТВЕННАЯ ПРОГРЕССИРУЮЩАЯ ПОЯСНО-КОНЕЧНОСТНАЯ МЫШЕЧНАЯ ДИСТРОФИЯ 2L ТИПА (АНОКТАМИНОПАТИЯ) // Современные проблемы науки и образования. 2020. № 4. ;URL: https://science-education.ru/en/article/view?id=29974 (дата обращения: 02.07.2026).
DOI: https://doi.org/10.17513/spno.29974