


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
COMPLEX MOLECULAR CHARACTERIZATION OF CERVICAL CANCER: METASTATIC MARKERS

Во всем мире ежегодно регистрируют 528 000 новых случаев рака шейки матки и 266 000 смертей от него, что превышает показатели любой другой гинекологической патологии [1]. 95% случаев вызваны персистирующими инфекциями ВПЧ [2]. Хотя ранние стадии рака шейки матки можно лечить хирургическим методом или лучевой терапией, метастатический рак шейки матки неизлечим, и необходимы новые терапевтические подходы [3].
Хотя большинство инфекций ВПЧ исчезают в течение месяцев, некоторые сохраняются и экспрессируют вирусные онкогены, которые инактивируют р53 и RB, что приводит к повышенной нестабильности генома, накоплению соматических мутаций и в некоторых случаях – к интеграции ВПЧ в геном хозяина [4, 5]. Связь с риском развития рака и гистологическими подтипами существенно различается среди канцерогенных типов ВПЧ, но причины этих различий плохо изучены. Кроме того, клинически значимые подгруппы пациентов с раком шейки матки еще предстоит определить. Целью данного исследования является всесторонний анализ баз данных и литературы по инвазивному раку шейки матки с акцентом на выявление клинических и молекулярных ассоциаций, а также функционально измененных сигнальных путей, которые могут стимулировать онкогенез и метастазирование и служить прогностическими или терапевтическими маркерами.
Соматические геномные изменения
С использованием базы данных TCGA было проанализировано 337 образцов опухолей шейки матки. В совокупности образцы содержали 43 324 соматические мутации, в том числе 24 551 миссенс, 2470 нонсенс, 9 260 сайленс, 5 841 некодирующую, 535 сплайс-сайтов, 74 нон-стоп мутации, 475 вставок и делеций со смещением рамки считывания (Indels) и 118 инсерций в рамке считывания. Одиннадцать опухолей с наибольшей частотой мутаций (>600 на образец) были классифицированы нами как «гипермутанты». Совокупная плотность мутаций составила 4,04 мутации на 1 Мб во всех опухолях и 2,53 после исключения опухолей-«гипермутантов».
Четырнадцать генов, в которых мутации встречались с наибольшей частотой (FDR <0,1), были выделены с использованием алгоритма MutSig2CV. Мы определили SHKBP1, ErbB3, Casp8, HLA-A и TGFBR2 в качестве новых статистически значимо мутированных генов (СМГ) при раке шейки матки, а также подтвердили, что PIK3CA, EP300, FBXW7, HLA-B, PTEN, NFE2L2, ARID1A, KRAS и MAPK1 являются статистически значимо мутированными, как было показано ранее [6, 7] (рис. 1).
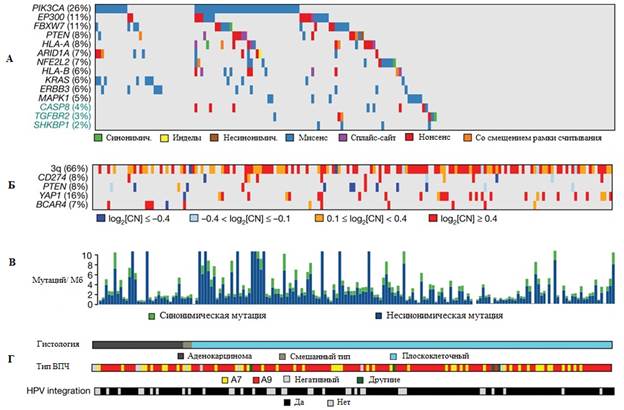
Рис. 1. Соматические геномные изменения при раке шейки матки:
А – гены со статистически значимыми мутациями, Б – изменения количества копий гена,
В – образцы рака шейки матки, упорядоченные по гистологии и частоте мутаций,
Г – клинические особенности исследованных образцов
В таблице 1 показано сравнение СМГ, идентифицированных на основе данных TCGA и ранее опубликованных данных [7]. Мутации в 7 из 14 СМГ (по данных TCGA) присутствовали по меньшей мере в одной плоскоклеточной карциноме и одной аденокарциноме. Однако мутации в HLA-A, HLA-B, NFE2L2, MAPK1, Casp8, SHKBP1 и TGFBR2 были найдены исключительно в плоскоклеточных опухолях.
В гене PIK3CA описаны в основном активирующие мутации E542K и E545K. Эти данные схожи с результатами, полученными по раку мочевого пузыря [8] и ВПЧ-позитивному плоскоклеточному раку головы и шеи (HNSCs), но отличаются от данных по раку молочной железы и большинству других видов рака [9].
Таблица 1
Результаты MUTSIG2CV
|
Данные TCGA (без гипермутантов) (n=181) |
Данные Ojesina et al. [7](n=115) |
|||
|
Ген |
q-value |
Ген |
q-value |
|
|
FBXW7 |
1.02E-12 |
FBXW7 |
7.16E-11 |
|
|
KRAS |
1.02E-12 |
KRAS |
0.00347 |
|
|
MAPK1 |
3.69E-08 |
MAPK1 |
0.00304 |
|
|
PTEN |
1.17E-06 |
PTEN |
3.29E-05 |
|
|
HLA-A |
4.38E-06 |
HLA-A |
0.439 |
|
|
HLA-B |
2.95E-05 |
HLA-B |
0.0065 |
|
|
ARID1A |
4.76E-04 |
ARID1A |
1.00E+00 |
|
|
EP300 |
1.35E-03 |
EP300 |
0.00418 |
|
|
PIK3CA |
2.92E-03 |
PIK3CA |
<1.82e-11 |
|
|
NFE2L2 |
6.41E-03 |
NFE2L2 |
0.14 |
|
|
CASP8 |
3.13E-02 |
CASP8 |
0.141 |
|
|
ERBB3 |
5.80E-02 |
ERBB3 |
1.00E+00 |
|
|
STK11 |
1.00E+00 |
STK11 |
0.138 |
|
|
Данные TCGA (без гипермутантов) |
Данные Ojesina et al. [7] |
|||
|
Плоскоклеточный рак (n=148) |
Плоскоклеточный рак (n=79) |
|||
|
Ген |
q-value |
Ген |
q-value |
|
|
FBXW7 |
2.70E-08 |
FBXW7 |
4.03E-12 |
|
|
MAPK1 |
3.64E-08 |
MAPK1 |
6.71E-04 |
|
|
HLA-A |
1.84E-06 |
HLA-A |
2.81E-01 |
|
|
HLA-B |
1.24E-05 |
HLA-B |
1.69E-03 |
|
|
PTEN |
2.23E-04 |
PTEN |
6.93E-02 |
|
|
NFE2L2 |
2.85E-03 |
NFE2L2 |
5.97E-02 |
|
|
EP300 |
5.51E-03 |
EP300 |
3.54E-02 |
|
|
PIK3CA |
6.92E-03 |
PIK3CA |
9.08E-12 |
|
|
CASP8 |
2.68E-02 |
CASP8 |
1.10E-01 |
|
|
ERBB3 |
7.22E-02 |
ERBB3 |
1.00E+00 |
|
|
KRAS |
7.22E-02 |
KRAS |
1.00E+00 |
|
|
TGFBR2 |
7.22E-02 |
TGFBR2 |
1.00E+00 |
|
|
SHKBP1 |
8.45E-02 |
SHKBP1 |
1.00E+00 |
|
|
STK11 |
1.00E+00 |
STK11 |
1.20E-02 |
|
|
Данные TCGA (без гипермутантов) |
Данные Ojesina et al. [7] |
|||
|
Аденокарцинома (n=30) |
Аденокарцинома (n=24) |
|||
|
Ген |
q-value |
Ген |
q-value |
|
|
KRAS |
1.38E-09 |
KRAS |
1.31E-01 |
|
|
ARID1A |
8.78E-04 |
ARID1A |
9.38E-01 |
|
|
CBFB |
1.00E+00 |
CBFB |
3.42E-02 |
|
|
ELF3 |
1.00E+00 |
ELF3 |
3.00E-02 |
|
Важной молекулярной характеристикой любого типа опухолей является показатель копийности генов (Copy Number Variation (CNV)). CNV – вид генетического полиморфизма, результатом которого может явиться снижение или повышение числа копий определенного гена и, следовательно, пониженная или повышенная экспрессия продукта гена – белка или некодирующей РНК [10-12].
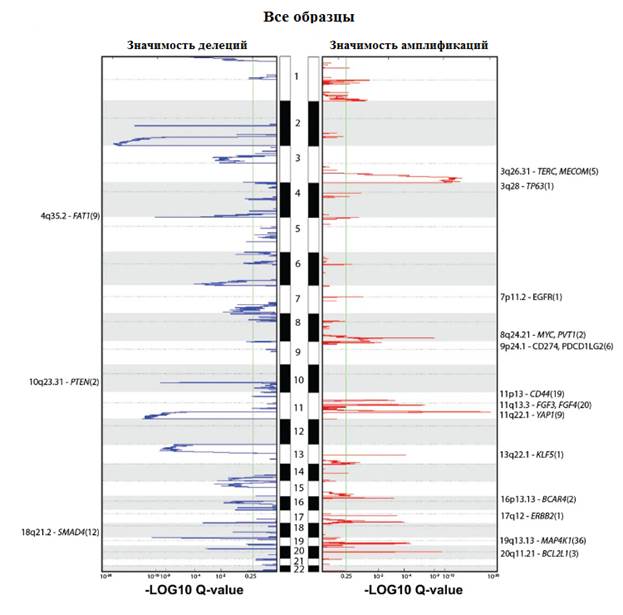
Рис. 2. Хромосомная локализация статистически значимых амплификаций (красный) и делеций (синий) генов, значения представлены по шкале -log10 q для всех образцов TCGA. Пики аннотированы драйверными генами. Общее количество генов в области пика указано в скобках. Аннотированные гены имеют значительную положительную корреляцию с экспрессией, соответствующей мРНК
Проанализировав данные TCGA, мы обнаружили в среднем на опухоль изменение копийности 88 генов, что меньше, чем при HNSC и раке яичников [13, 14, 15]. Анализ GISTIC2.0 выявил 26 амплификаций генов и 37 делеций (рис. 2). Амплификации были идентифицированы в участках 7p11.2 (EGFR, 17%), 9p24.1 (CD274, PDCD1LG2, 21%), 13q22.1 (KLF5, 18%) и 16p13.13 (BCAR, 420%), 3q26.31 (TERC, MECOM, 78%), 3q28 (TP63, 77%), 8q24.21 (MYC, PVT1, 42%), 11q22.1 (YAP1, BIRC2, BIRC3, 17%) и 17q12 (ERBB2, 17%).
Делеции были идентифицированы в 3p24.1 (TGFBR2, 36%) и 18q21.2 (SMAD4, 28%), 4q35.2 (FAT1, 36%) и 10q23.31 (PTEN, 31%). Кластер с большими изменениями числа копий генов в основном содержал плоскоклеточные опухоли с амплификациями участков 11q22 (YAP1, BIRC2, BIRC3) и 7p11.2 (EGFR), в то время как кластер с низкой частотой изменения числа копий генов включал большинство аденокарцином и был представлен опухолями с делециями TGFBR2 и SMAD4, а также амплификациями ERBB2 и KLF5 (рис. 3).
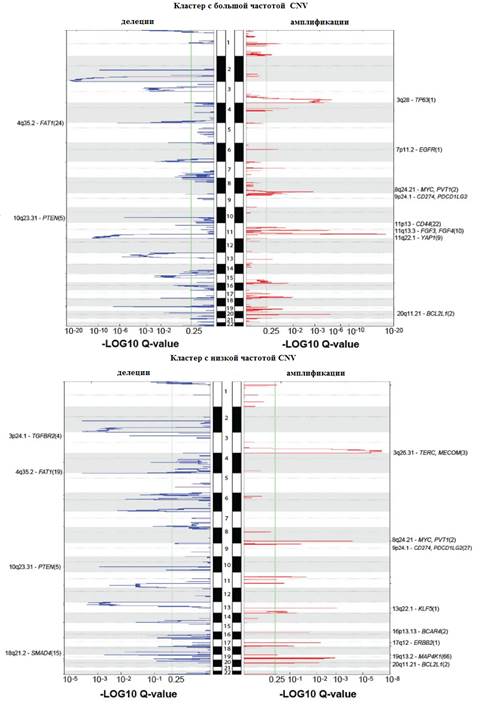
Рис. 3. Графики амплификаций и делеций генов по CNV-кластерам (получены с помощью модуля GISTIC2.0)
Примечательно, что обе группы имели амплификации с участием CD274 (PD-L1) и PDCD1LG2 (PD-L2), которые статистически значимо коррелировали (P <0,0001) с экспрессией двух ключевых иммунных цитолитических эффекторных генов – гранзима A и перфорина [16]. Это подчеркивает потенциал иммунотерапевтических стратегий для некоторых видов рака шейки матки.
Структурные перестройки были идентифицированы с помощью данных секвенирования РНК (RNA-seq) (TCGA, n=78) и данных секвенирования всего генома (WGS). Как RNA-seq, так и WGS выявили 22 структурные перестройки у 14 пациентов. В общей сложности было обнаружено 26 слияний (рис. 4). Анализ данных RNA-seq выявил четыре образца с 16p13 слияниями гена ZC3H7A – BCAR4, в результате чего экзон 1 ZC3H7A был связан с последним экзоном BCAR4. WGS обнаружил тандемную дупликацию и увеличение числа копий BCAR4 на хромосоме 16p13.13.
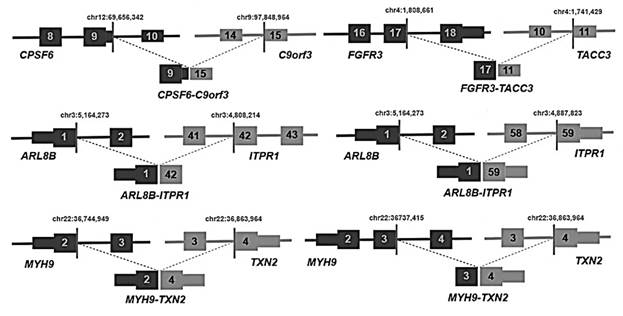
Рис. 4. Схема слияния CPSF6-C9orf3, ARL8B-ITPR1 и MYH9-TXN2, а также слияний с известными проявлениями при других типах рака (FGFR3-TACC3), обнаруженных по меньшей мере в двух исследованиях RNA-seq
BCAR4 – это длинная некодирующая РНК, способствующая метастазированию, которая усиливает пролиферацию клеток при резистентном к эстрогену раке молочной железы, активируя путь HER2/HER3. Лапатиниб, ингибитор EGFR / HER2, противодействует росту опухоли, вызванной BCAR4 in vitro, что требует дальнейших исследований для оценки потенциала этого терапевтического средства при BCAR4-позитивном раке шейки матки [17].
Комплексный анализ молекулярных подгрупп рака шейки матки
Интеграция данных о копийности генов, метилировании, экспрессии мРНК и микроРНК с использованием iCluster [18] показала молекулярную гетерогенность рака шейки матки. Были идентифицированы 3 кластера, которые в основном соответствуют кластерам экспрессии мРНК: кластер (к-1) – плоскоклеточный рак с высокой экспрессией семейства кератиновых генов, кластер (к-2) – плоскоклеточный рак с более низкой экспрессией генов кератина и кластер аденокарцином (к-3). Мутации в генах KRAS (P =9,7 × 10-5), ERBB3 (P =2,6 × 10 -3) и HLA-A (P=0,03) были связаны с кластерами, при этом мутации KRAS отсутствовали в кластере с высоким уровнем экспрессии генов кератина, а мутации HLA-A отсутствовали в кластере аденокарцином (рис. 5). Гены ARID1A (P =0,02), NFE2L2 (P =6,9 × 10-6) и PIK3CA (P =0,01) были дифференциально экспрессированы в кластерах 1 и 2.
Иерархическая кластеризация вариабельных данных по оценке уровня ДНК-метилирования позволила выделить три группы, включая кластер CpG-островков с гиперметилированием (CIMP(CpG island methylator phenotype)-high), кластер с промежуточным уровнем CIMP и кластер с низким уровнем CIMP, которые были связаны с уровнем мРНК генов эпителиально-мезенхимального перехода (EMT) [19].
Большинство образцов в кластере аденокарциномы имели высокий уровень CIMP, тогда как другие группы iCluster содержали образцы с промежуточным и низким уровнем CIMP (рис. 5). Сравнивая все опухоли шейки матки с нормальными образцами, взятыми из 12 проектов TCGA, мы идентифицировали 1026 эпигенетически подавленных генов, включая ZNF, ADAM, ADAMTS и COL, которые были метилированы в большей степени при раке, чем в нормальных тканях.
Кластеризация по уровню экспрессии микроРНК позволила выделить 6 кластеров (P =1,7 × 10 -19). Образцы из кластера аденокарцином перекрывались с кластером № 5 miRNA и характеризовались высокой экспрессией miR-375 и низкой экспрессией miR-205-5p и miR-944 (рис. 6).
Уровни экспрессии опухолевых супрессоров miR-99a-5p и miR-203a были значительно выше в образцах кластера с высоким уровнем экспрессии генов кератина, чем в образцах кластера с низким уровнем экспрессии генов кератина (P =0,01 и P=0,008 соответственно).
Среди miRNAs со значительными и функционально подтвержденными антикорреляциями 22 генов и белков выделяются микроРНК семейства miR-200, которые коррелировали с EMT-связанными факторами транскрипции ZEB1, ZEB2 и SNAI2, геном YAP1, ERBB2, ERBB3 и AXL и геном рецептора эстрогена ESR1. Анализ белковых RPPA (reverse phase protein lysate microarray)-микрочипов со 192 антителами (рис. 6) выявил 3 кластера, достоверно связанных с группами iCluster (P=1,8 × 10 -4) и экспрессий мРНК EMT. Образцы из кластера EMT были связаны с кластером плоскоклеточного рака с низким уровнем экспрессии кератина, тогда как образцы PI3K-AKT и гормонального кластера были связаны с кластерами плоскоклеточного рака с высоким уровнем кератина и аденокарциномой, что указывает на активацию различных сигнальных путей в этих подтипах рака шейки матки.
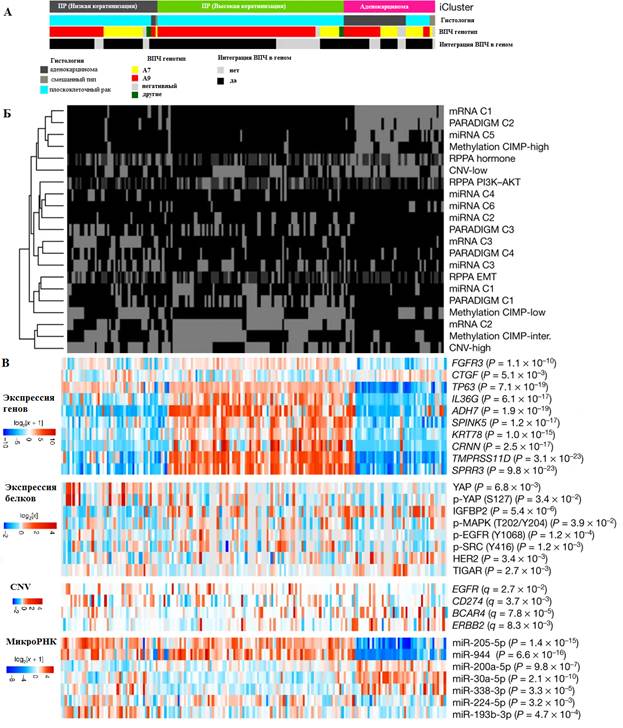
Рис. 5. Мультиплатформенная интегративная кластеризация опухолей шейки матки:
А. Интегративная кластеризация 178 образцов цервикального рака с использованием данных мРНК, метилирования, микроРНК и вариации числа копий (CNV) идентифицирует две группы плоскоклеточного рака (с низкой и высокой экспрессией генов кератина) и одну группу аденокарциномы. Представленные характеристики включают гистологию, статус ВПЧ и статус интеграции ВПЧ, UCEC-подобный статус, уровень мутагенеза APOBEC, показатель мРНК EMT, чистоту опухоли и три SMG (KRAS, ERBB3 и HLA-A), которые значительно связаны между тремя кластерами, идентифицированными с помощью iCluster.
Б. Тепловая карта кластеров (данные по мРНК, микроРНК, метилированию, CNV и PARADIGM). C1 – C6 обозначают кластеры.
В. Тепловые карты показывают выбранные мРНК, микроРНК, белки и CNV, которые либо значительно связаны с группами iCluster, либо были идентифицированы как маркеры в других анализах
Дифференциальные уровни экспрессии фосфорилированных белков p-MAPK, p-EGFR (Y1068), p-SRC (Y416), IGFBP2 и TIGAR в двух кластерах плоскоклеточного рака предполагают различные паттерны активации RTK, MAPK, PI3K и метаболических сигнальных путей, которые могут лежать в основе молекулярного разнообразия плоскоклеточного рака шейки матки (рис. 6).
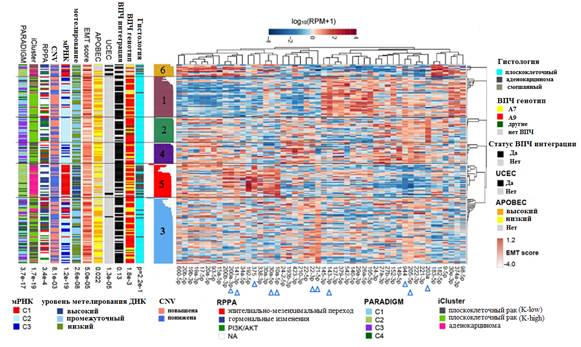
Рис. 6. Кластеры микроРНК в 178 образцах опухолей шейки матки (данные SAMseq) и их корреляционные взаимосвязи между генами и белками. Синими треугольниками отмечены микроРНК, ассоциированные с раком, дифференциально экспрессированные при различных гистологических подтипах
Основные члены каждого кластера RPPA были связаны с пятилетней выживаемостью, причем группа EMT показала худший результат. Примечательно, что это была единственная платформа (протеомное профилирование), на которой кластеры связаны с выживаемостью. Образцы в кластере ЕМТ показали наличие подмножества стромальных опухолей, которые имеют высокую экспрессию MYH11 и RAB11. YAP был наиболее сильно экспрессированным белком, отличающим образцы кластера EMT от всех других, и ген YAP1 также был гиперэкспрессирован в образцах из кластера EMT по сравнению с кластером PI3K-AKT. Регулирование связанных с EMT молекул YAP и ZEB1 [20, 21, 22] также может быть обусловлено значительно более низкими уровнями экспрессии miR-200a-3p в образцах из кластера EMT по сравнению с образцами из других кластеров RPPA. Эти результаты подчеркивают потенциальную роль YAP в EMT-регулируемой прогрессии рака шейки матки.
Алгоритм модулей взаимной исключительности (MEMo) [23] использует данные соматических мутаций и числа копий гена для идентификации онкогенных сетей с взаимоисключающими геномными изменениями. Поскольку экспрессия miR-200a и miR-200b (miR-200a / b) отрицательно коррелировала с оценками мРНК EMT, Gregory P. и соавторы использовали MEMo для изучения изменений в генных сетях miR-200a/b и EMT и обнаружили потенциальную связь между путем TGFβ и изменениями miR-200a/b в регуляции EMT [24, 25]. Делеции и мутации, затрагивающие ген рецептора TGFBR2, модулирующие гены CREBBP и EP300 и фактор транскрипции SMAD, вероятно, влияют на рост-подавляющие и проапоптотические функции (рис. 7) и наблюдались в 30% плоскоклеточных карцином.
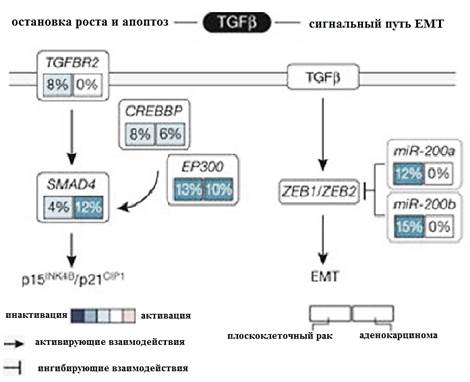
Рис. 7. Взаимоисключительность соматических изменений в сигнальном пути TGFβR2. (Сигналы TGFβ часто изменяются в опухолях шейки матки. Изменения в этом пути делятся между теми, которые, вероятно, влияют на функции подавления опухоли TGFβ, и теми, которые влияют на программу EMT, управляемую TGFβ.)
Плоскоклеточные опухоли с низкой экспрессией генов кератина как с гиперметилированием, так и с пониженной регуляцией miR-200a / b показали значительную активацию как ZEB1, так и ZEB2 и были взаимоисключающими с изменениями в сигнальном пути TGFβ. Примечательно, что образцы с измененным miR-200a / b показали более высокие оценки мРНК EMT, чем неизмененные образцы, тогда как не было обнаружено существенного различия между образцами с изменениями TGFβ-пути или без них. Эти результаты подчеркивают потенциальные подходы к лечению этой подгруппы пациентов с раком шейки матки, поскольку нацеливание на EMT может сделать опухоли более чувствительными к низкомолекулярным ингибиторам и цитотоксической химиотерапии [19, 26, 27].
Анализ MEMo также показал различия в терапевтически значимых изменениях путей RTK, PI3K и MAPK при раке шейки матки. Обнаружена сильная тенденция к совместному появлению изменений ERBB2 и ERBB3 в аденокарциномах, что указывает на то, что подгруппа этих опухолей может демонстрировать аберрантную передачу сигналов HER3 через взаимодействия между мутантом HER3 и активированным HER2 и, следовательно, способна потенциально обеспечить преимущество HER2- и HER3-ориентированной терапии [28]. Аберрации в PIK3CA также имеют тенденцию сочетаться с соматическими мутациями и делециями PTEN, что аналогично опухолям эндометрия с небольшим изменением числа копий и предполагает потенциальную терапевтическую пользу от PI3K-таргетных препаратов [14].
PARADIGM [29, 30], которая объединяет копийность генов, данные RNA-seq и взаимодействие сигнальных путей, показал заметно отличающиеся молекулярные профили между плоскоклеточным раком и аденокарциномой. PARADIGM идентифицировал более высокую предполагаемую активацию сигнальных путей p53, p63, p73, AP-1, MYC, HIF1A, FGFR3 и MAPK в качестве ключевых отличительных признаков плоскоклеточного рака, сходного с другими плоскоклеточными раками [31]. В отличие от этого, аденокарциномы демонстрировали более высокую активацию путей ERα. Ведущую роль в патогенезе данного заболевания играет гиперэстрогения. Эстрогены стимулируют пролиферацию клеток, индуцируя синтез факторов роста и их рецепторов, в том числе и эстрогеновый рецептор α типа (ERα или ESR1) [32]. Возможные механизмы, лежащие в основе повышающей активности ERα, могут проистекать из экспрессии miR-193b-3p, прямого регулятора ESR1, который был значительно подавлен при аденокарциномах по сравнению с плоскоклеточным раком, или от передачи сигналов эстрогена в стромальных клетках [33].
Генотипы и интеграция ВПЧ
Из 178 опухолей 169 (95%) были ВПЧ-положительными, 120 (67%) имели альфа-9 (А9) типы (103 ВПЧ16), 45 (25%) имели альфа-7 (А7) типы (27 HPV18) и 9 (5%) были HPV-отрицательными. Варианты ВПЧ были преимущественно европейскими (137 из 169, 81% вариантов А), и была отмечена значительная связь неевропейских вариантов ВПЧ16 с аденокарциномами. Все HPV-позитивные виды рака имели детектируемую экспрессию мРНК E6- и E7-онкогена HPV, которые кодируют белки, ингибирующие функции р53 и RB соответственно [5, 34, 35]. Примечательно, что HPV18 опухоли имели значительно более высокие соотношения несплайсированных и сплайсированных транскриптов, кодирующих активный онкопротеин E6, чем другие опухоли, что указывает на различные функциональные значения E6 и E7 в опухолях, связанных с разными генотипами ВПЧ.
Типы ВПЧ A7 были представлены в кластерах с плоскоклеточным раком и аденокарциномами. Большинство опухолей ВПЧ A7 были слабо метилированы (CIMP-low), а HPV-отрицательные опухоли образовывали отчетливую подгруппу в кластере CIMP-low со значительно более низким средним уровнем промотора-метилирования, чем в других образцах в этом кластере. Образцы с самой высокой степенью сайленсинга генов были ВПЧ-положительными аденокарциномами (генотип A9). Функциональный эпигенетический модуль анализа, который объединяет данные ДНК-метилирования и экспрессии генов с использованием сетей взаимодействий, выявил обратные корреляции между метилированием и экспрессией генов при ВПЧ-положительном и ВПЧ-отрицательном раке шейки матки. Анализ выявил 12 статистически значимых подсетей для рака шейки матки. miR-944, miR-767-5p и miR-105-5p были наиболее дифференцированно экспрессируемыми miRNAs между HPV-положительными и HPV-отрицательными образцами. Экспрессия miR-944 также была значительно выше, в то время как экспрессия miR-375 была значительно ниже при плоскоклеточном раке с HPV16 по сравнению с плоскоклеточным раком с HPV18. Примечательно, что HPV-негативные виды рака имели значительно более высокий показатель мРНК EMT и более низкую частоту сигнатуры мутагенеза APOBEC по сравнению с HPV-позитивными опухолями [36].
PARADIGM использовали для оценки молекулярных путей, дифференциально активированных в плоскоклеточных опухолях с инфекциями HPV генотипов A7 и A9. Наблюдалась более высокая активация сигнальных путей p53 и p63 и более низкая – сигнального пути FOXA1 в опухолях, инфицированных типами A9 (рис. 8). Более высокая активация пути SFN также наблюдалась при A9-позитивных опухолях, что согласуется с низким метилированием и высоким уровнем экспрессии генов SFN, обнаруженным при функциональном анализе эпигенетического модуля. Примечательно, что SFN белок (также известный как 14-3-3σ) ранее был связан с эпителиальной иммортализацией и плоскоклеточным раком, измененной активацией сигнального пути р53 и Wnt-опосредованной передачей сигналов β-катенином [37, 38].
Вирусно-клеточные транскрипты, указывающие на интеграцию ВПЧ в геном хозяина, наблюдались в 141 из 169 (83%) ВПЧ-позитивных опухолей, включая все ВПЧ-18-позитивные формы. Из этих 141 опухоли 90 (64%) имели одно событие интеграции ВПЧ, 35 имели два события и 16 имели три или более событий (всего 220 уникальных событий интеграции). События интеграции ВПЧ повлияли на все хромосомы, включая некоторые ранее описанные горячие точки, такие как 3q28 и 8q24 (рис. 8).
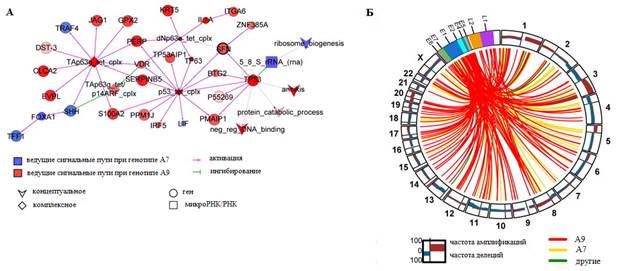
Рис. 8. Интеграция ВПЧ и активация сигнальных путей, дифференциальных между подтипами ВПЧ:
A. Результат моделирования в Cytoscape самой большой взаимосвязанной регуляторной сети с функциями интегрированного уровня пути (integrated pathway levels, IPL) PARADIGM, показывающий дифференциальную активацию сигнальных путей при плоскоклеточном раке c HPV A9 и A7. Цвет узла и интенсивность отражают уровень дифференциальной активации. Размер узла представляет уровень значимости.
Б. Диаграмма Circos, показывающая частоту (0–100%) амплификаций и делеций для областей каждой хромосомы (внешнего круга). Линии во внутреннем круге указывают на точки интеграции генома ВПЧ (гены L1, L2, E1, E2, E4, E5, E6 и E7) в геном человека
Геномные локусы, затронутые интеграцией, характеризовались повышенными изменениями числа копий генов и повышенной экспрессией генов. Кроме того, 153 (70%) слитых транскриптов включали известные или предсказанные гены, тогда как остальные включали межгенные области [39].
Молекулярные механизмы и маркеры регионарного метастазирования при раке шейки матки
Общая выживаемость обычно высока среди пациентов с ранними стадиями рака шейки матки; тем не менее наличие метастазов в лимфатических узлах в четыре раза снижает выживаемость [40]. Молекулярные маркеры как средство выявления метастазов в лимфатических узлах могут иметь важное терапевтическое значение. Одним из таких маркеров может выступать изменение копийности генов (CNV). CNV представляет собой критическое генетическое событие, которое способствует развитию и прогрессированию (метастазированию) злокачественных новообразований у человека [41, 42].
Как описано выше, геномные изменения при раке шейки матки, в частности амплификации в хромосомах 1q, 3q и 5p, были зарегистрированы во многих исследованиях [43]. Потенциальные гены, представляющие интерес в этих областях амплификации, включают LAMP3, CCND1, PROX1 и PRKAA1. LAMP3 находится в области, имеющей большое значение для канцерогенеза шейки матки. Высокая экспрессия этого гена связана с повышенным метастатическим потенциалом при раке шейки матки. Ген PROX1 является специфическим маркером лимфатического эндотелия, участвующим в регуляции развития лимфатической системы. PRKAA1, клеточный регулятор метаболического стресса, может способствовать росту опухолевых клеток в условиях стресса и является потенциальным маркером канцерогенеза шейки матки [40]. CCND1 – генетический маркер клеточной пролиферации. Сверхэкспрессия CCND1 также связана с метастазами в лимфатические узлы [44].
В работе Wangsa D. и соавторов [40] обнаружено, что анализ числа геномных копий LAMP3, PROX1 и PRKAA1 позволяет статистически значимо прогнозировать метастазы в лимфатические узлы для рака шейки матки. Также в этой работе обнаружено, что клональные паттерны могут различаться между первичной опухолью и ее метастазированием, что указывает на то, что определенные клоны способны иметь преимущество при росте в среде лимфатического узла. До этого ни одно другое исследование не проводило анализы FISH для прогнозирования метастазов в лимфатических узлах при раке шейки матки. В исследованиях, направленных на прогнозирование метастазов в лимфатических узлах, использовались такие методы, как иммуногистохимия, ОТ-ПЦР и профилирование экспрессии генов. Иммуногистохимия является экономически эффективным методом, но для прогнозирования метастазов в лимфатических узлах при раке шейки матки сообщалось о различных эпитопах (Cox-2, p16, CXCR4, CCR7, D2-40 и т.д.) и противоречивых результатах [40]. Исследования профилирования экспрессии генов также обнаружили расходящиеся результаты в исследовании Biewenga et al. [45], который не смог предсказать метастазы в лимфатические узлы на ранней стадии рака шейки матки, в то время как в исследовании Kim et al [46] это было сделано. Biewenga et al. [45] обнаружил 5 генов, демонстрирующих дифференциальную экспрессию между пациентами с метастазами в лимфатических узлах и без них. Прогнозирование метастатического поражения лимфатических узлов поможет в разработке адекватных вариантов лечения для пациентов, поскольку гистологически необнаружимые микрометастазы в лимфатической системе могут быть причиной рецидива рака шейки матки. Гистологические исследования не являются непогрешимыми, так как включают только небольшую часть лимфатических узлов, а исследователь имеет шанс 1% обнаружить микрометастатический очаг в пределах диаметра трех опухолевых клеток [40].
Амплификация генов является распространенным механизмом активации онкогена, и при раке шейки матки часто наблюдается увеличение количества хромосом 1q, 3q и 5p [43]. В исследовании Wangsa D. и соавторов [40] LAMP3 амплифицирован, что подтверждает прирост числа копий в области 3q. Амплификация хромосомы 3q также была связана с метастазами в лимфатических узлах в двух исследованиях с использованием сравнительной геномной гибридизации, которая показала увеличение прироста числа копий 3q в первичных опухолях с метастазированием в лимфатические узлы; однако результаты не были статистически значимыми. Хотя функция LAMP3 полностью не известна, предполагают, что LAMP3 может быть вовлечен в миграцию опухолевых клеток в окружающие лимфатические сосуды. PROX1 был обнаружен исключительно в лимфатических эндотелиальных клетках и жизненно важен для развития эмбриональных лимфатических узлов [47]. Снижение экспрессии PROX1 было обнаружено при нескольких видах рака, включая гепатоцеллюлярную карциному, рак желчных протоков и рак молочной железы. Однако при раке толстой кишки избыточная экспрессия PROX1 вызывает прогрессирование рака толстой кишки, способствуя переходу от доброкачественного к высокодиспластическому фенотипу. При раке шейки матки PROX1 находится в области амплификации (хромосома 1q), что может указывать на то, что прирост числа копий PROX1 может функционировать аналогично таковому при раке толстой кишки. Развитие рака шейки матки также связано с увеличением числа копий в области хромосомы 5p, причем PRKAA1 является потенциально интересным геном. Так, показано, что высокая экспрессия PRKAA1 может помогать злокачественным опухолям поддерживать устойчивость к гипоксическому состоянию [48].
Клональные паттерны могут варьировать среди первичных опухолей шейки матки. Исследование Wangsa D. и соавторов [40] – это первое исследование, изучающее разницу в паттернах маркеров между клетками первичного рака шейки матки и синхронными метастазами в лимфатических узлах. В этой работе несколько случаев показали одинаковые клоны и их частоту между первичной опухолью и метастазами в лимфатических узлах, что указывает на то, что основной клон(ы) первичной опухоли были также наиболее успешными клонами для формирования метастазов в лимфатических узлах. Следовательно, в этих случаях, по крайней мере на тестируемом геномном уровне, большинство клеток первичной опухоли могут стать метастатическими клетками. В этом случае метастатический потенциал уже будет присутствовать в объеме опухоли. Однако в некоторых случаях изменения в клональной конституции между первичной опухолью и метастазами в лимфатических узлах указывают на то, что ранее существовавший, но незначительный клон в первичной опухоли имел преимущества для развития метастазов в лимфатических узлах по сравнению с основным клоном первичной опухоли. Это соответствует гипотезе о том, что подмножество клеток в первичной опухоли имеет генетическую структуру, которая в конечном итоге приводит к метастазированию, что может указывать на то, что для метастазирования необходимы дополнительные мутации в нескольких клетках. Это различие в паттернах может отражать различные механизмы развития метастазов в лимфатических узлах при раке шейки матки.
В исследовании Zhou Y и соавторов [49] экспрессия WNT2 часто наблюдалась в образцах с метастазами в периферические лимфатические узлы (PLNM) по сравнению с экспрессией в образцах без PLNM и, таким образом, выделялась в качестве потенциального гена-кандидата в регуляции PLNM. Семейство генов WNT человека состоит из 19 членов, которые кодируют эволюционно консервативные гликопротеины. Передача сигналов WNT участвует в регуляции пролиферации, дифференцировки, апоптоза и миграции клеток. В последнее десятилетие раскрыта роль WNT2 в развитии и прогрессировании различных видов рака. Так, WNT2 активируется при раке желудка и может влиять на образование, инвазию и распространение опухоли. Опухолевые фибробластные клетки также секретируют WNT2, который действует как фактор, способствующий росту и инвазии, активируя канонический сигнальный путь WNT / β-catenin в раковых клетках пищевода [49].
В исследовании Zhou Y и соавторов уровень экспрессии WNT2 (как мРНК, так и белка) повышен при раке шейки матки по сравнению с неопухолевыми тканями шейки матки. Наблюдалась значительная связь между высокой экспрессией WNT2 и размером опухоли. Важно, что уровни экспрессии WNT2 и PLNM были независимыми прогностическими факторами для плохих показателей выживаемости у пациентов на ранней стадии рака шейки матки. Более того, нокдаун WNT2 приводит к снижению подвижности клеток [49]. Учитывая это, можно считать, что WNT2 является потенциальным новым предиктором PLNM и многообещающей терапевтической мишенью при раке шейки матки.
До сих пор стандартное лечение ранней стадии рака шейки матки включало радикальную гистерэктомию плюс лимфаденэктомию или химиорадиотерапию, при которых имеются сходные показатели выживаемости. Химиолучевая терапия необходима для пациентов с метастазами в лимфатических узлах, что делает начальную хирургическую процедуру ненужной в ретроспективе. Тем не менее, поскольку невозможно точно и эффективно клинически определить метастазы в тазовые и парааортальные лимфатические узлы, онкологи не могут определять стратегию лечения, избегать ненужного хирургического вмешательства [49].
Точность прогнозирования PLNM путем определения экспрессии WNT2 (высокая или низкая) составляет 36%. Поскольку сообщаемый показатель микрометастаза в лимфатических узлах тазовых органов составляет 15% у пациентов с ранней стадией рака шейки матки, весьма вероятно, что фактическая точность прогнозирования PLNM путем определения экспрессии WNT2 может составлять 51% [40].
Заключение
Благодаря комплексному молекулярному профилированию в настоящее время выявлены новые геномные и протеомные характеристики, которые подразделяют рак шейки матки на несколько подтипов. Кластерный анализ идентифицировал слабокератинизированные плоскоклеточные опухоли, кератинизированные плоскоклеточные опухоли и аденокарциномы, отличающиеся по ВПЧ-статусу и молекулярным особенностям. ErbB3, Casp8, HLA-А, SHKBP1 и TGFBR2 были идентифицированы как гены с наиболее сильно выраженными мутационными изменениями при первичном раке шейки матки, а ErbB3 (HER3) – непосредственно как терапевтическая мишень. Кроме того, идентифицированы амплификации в генах CD274 и PDCD1LG2, которые кодируют хорошо известные мишени для иммунотерапии. Был обнаружен ряд эндометриально-подобных раковых заболеваний шейки матки, состоящих преимущественно из ВПЧ-негативных опухолей и характеризующихся мутациями в KRAS, ARID1A и PTEN, при этом белки PTEN и, возможно, ARID1A могут служить терапевтическими мишенями. Важно отметить, что более чем в 70% случаев рака шейки матки отмечаются геномные изменения в одном или обоих сигнальных путях PI3K-MAPK и TGFβ, что иллюстрирует потенциальную клиническую значимость терапевтических агентов, нацеленных на участников этих путей. Вместе эти результаты дают представление о молекулярных подтипах рака шейки матки и служат обоснованием необходимости разработки различных методов лечения для этих подтипов.
В настоящее время невозможно точно и клинически эффективно указать метастазы в тазовые и парааортальные лимфатические узлы при раке шейки матки, поэтому онкологи не могут точно определять стратегию лечения и избежать ненужного хирургического вмешательства. Однако результаты исследований последнего десятилетия свидетельствуют, что показатель копийности генов LAMP3, PROX1, PRKAA1 и WNT2 является потенциальным новым предиктором метастазирования в лимфатические узлы, а сами гены – многообещающей терапевтической мишенью при раке шейки матки.
Библиографическая ссылка
Кечерюкова М.М., Снежко А.В., Вереникина Е.В., Меньшенина А.П., Адамян М.Л., Арджа А.Ю., Кечерюкова Т.М. КОМПЛЕКСНАЯ МОЛЕКУЛЯРНАЯ ХАРАКТЕРИСТИКА РАКА ШЕЙКИ МАТКИ: МАРКЕРЫ МЕТАСТАЗИРОВАНИЯ // Современные проблемы науки и образования. 2020. № 2. ;URL: https://science-education.ru/en/article/view?id=29769 (дата обращения: 27.07.2026).
DOI: https://doi.org/10.17513/spno.29769