


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
MOLECULAR CYTOGENETIC AND BIOINFORMATIC RESEARCH: DELETION OF CHROMOSOME 6 (6q22.1-q23.2) AND POTENTIAL OF INTERACTOME ANALYSIS

Частота умственной отсталости, аутизма, эпилепсии у детей широко варьирует и, по различным данным, может превышать 3% в общей популяции [1-5]. Этим заболеваниям нередко сопутствуют врожденные пороки и/или микроаномалии развития. В случае отсутствия экзогенных факторов, негативно влияющих на формирование центральной нервной системы (ЦНС), возникновение данного нарушения, как правило, связывают с генетическими нарушениями. При наличии признаков нервного и/или психического расстройств для выявления генетических факторов заболевания применяются различные методы диагностики. К последним можно отнести цитогенетический анализ и молекулярное кариотипирование (серийную сравнительную геномную гибридизацию - arrayCGH). Разрешающая способность цитогенетического исследования составляет 5 и более млн пн, в то время как молекулярное кариотипирование позволяет выявлять хромосомные микроаномалии размером от 500 пн [6-8]. В российских когортах детей с нарушением психики и пороками развития геномная патология выявляется примерно в половине случаев, из которых 8-12% определяют классическим кариотипированием и до 40% - молекулярно-цитогенетическими методами [9]. В остальных случаях причину патологии, как правило, выявить не удается. Это связано с тем, что нервные и психические заболевания могут быть связаны с генными мутациями, не выявляемыми указанными методами.
Интерпретация геномных аномалий, выявленных молекулярно-цитогенетическими методами, как правило, требует применения методов биоинформатики. Наследование многих психических заболеваний носит полигенный характер, а их проявления варьируют в зависимости от функций и числа затронутых генов в сочетании с индивидуальными генетическими особенностями. Помимо патогенных геномных нарушений, ассоциированных с различными заболеваниями, в популяции присутствует большое количество рекуррентных или часто встречающихся перестроек, считающихся непатогенными [10]. Однако если несколько рекуррентных нарушений затрагивают одну интерактомную сеть, то в результате кумулятивного эффекта они могут создать основу для развития заболевания. Данные молекулярного кариотипирования позволяют уточнить локализацию нарушения и идентифицировать гены, ассоциированные с обнаруженной микроаномалией. Затем, используя биоинформатический анализ, можно оценить вовлеченность в патогенез каждого гена, сделать предположение о процессах или каскаде процессов, лежащих в основе развития заболевания. Анализ фенотипических и патофизиологических нарушений способствует поиску эффективных способов коррекции нарушений, вызванных генетическими аномалиями.
В данной работе мы представляем клиническую, цитогенетическую и молекулярно-цитогенетическую характеристику делеции длинного плеча хромосомы 6 в участке 6q22.1q23.2 у ребенка 5 лет. Приведен детальный анализ генов, локализованных в области аномалии, с обсуждением корреляции генотип-фенотип.
Цель работы
Цель настоящей работы заключалась в описании молекулярно-цитогенетических и биоинформатических исследований хромосомной патологии у ребенка с умственной отсталостью и врожденными пороками развития на основе применения анализа геномных вариаций и их функциональных последствий.
Материалы и методы
Цитогенетическое исследование. Препараты метафазных хромосом получали путем фиксации лимфоцитов периферической крови, после культивирования in vitro в соответствии со стандартной методикой. Цитогенетический анализ проводился при помощи дифференциального окрашивания хромосом по длине (G- и C-окрашивание) по общепринятым протоколам [3] под световым микроскопом при увеличении х1125.
Молекулярно-цитогенетическое исследование. Подготовка ДНК и анализ методом молекулярного кариотипирования проводили согласно ранее описанному протоколу с использованием SNP/олигонуклеотидной микроматрицы c разрешением не менее 1 тысячи пн (Affymetrix) [3-5].
Биоинформатический анализ. Изучение транскриптомных, метаболомных и интерактомных данных проводили с использованием оригинальной биоинформатической технологии [11; 12]. Межбелковые взаимодействия оценивались с использованием ресурса Mentha [13].
Результаты и обсуждение
В лабораторию молекулярной цитогенетики нервно-психических заболеваний была направлена девочка 5 лет с задержкой психоречевого развития и эпизодами фебрильных судорог для исследования кариотипа. Ребенок родился от 2-й беременности, нормальных родов. Беременность протекала с гестозом в первом триместре, повышенным артериальным давлением и угрозой прерывания. Масса ребенка при рождении составила 2800 г, рост - 50 см. Раннее моторное развитие ребенка протекало с отставанием: сидеть стала с 9 месяцев, ходить с - 1 года 11 месяцев. Фразовая речь появилась с трех лет. Первый эпизод фебрильных судорог произошел в 1 год и 3 месяца. У пациентки есть старший брат, развивающийся нормально.
При обследовании врачом-генетиком у ребенка были выявлены следующие микроаномалии развития: долихоцефалия, тонкие редкие волосы, узкое лицо, гипотелоризм глазных щелей, увеличение средней части лица, ретрогнатия, диспластичные ушные раковины, мелкие дистрофичные зубы, тонкие ногти, брахидактилия. Отмечалась умеренная мышечная гипотония, кифоз в грудном отделе позвоночника. В анамнезе наблюдались хореиформные гиперкинезы в мышцах конечностей, лица, шеи. Ребенку был поставлен диагноз: задержка психоречевого развития, симптоматическая эпилепсия, гиперкинетический синдром и гиперактивность. При магнитно-резонансной томографии (МРТ) была выявлена субатрофическая вентрикуломегалия резидуального характера. Офтальмологом обнаружена ангиопатия сетчатки.
В результате проведенного цитогенетического анализа была обнаружена интерстициальная делеция в длинном плече хромосомы 6 с предполагаемыми точками разрыва (кариотип - 46,ХХ,del(6)(q22.?q23.?3) (рис. 1).
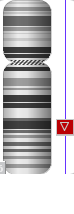

Рис. 1. Результаты цитогенетического исследования: обнаружена делеция хромосомы 6 в предполагаемом участке q22.?q23.?3
Для уточнения точек разрыва делеции и поиска других хромосомных микроперестроек было проведено молекулярно-цитогенетическое исследование методом молекулярного кариотипирования/SNParray. Исследование подтвердило наличие делеции, затрагивающей участок 6q22.1q23.2. Размер её составил 13,7 млн пн (геномные координаты: 117898555-131656086) (рис. 2). Других значимых (патогенных) геномных перестроек обнаружено не было.
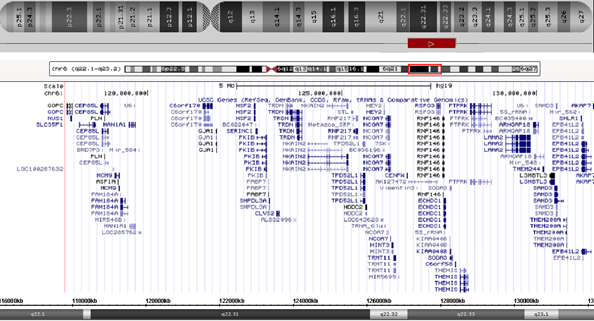
Рис. 2. Схематическое изображение делеции хромосомы 6, обнаруженной с помощью молекулярного кариотипирования
Согласно UCSC [14] в области делеции локализовано 66 генов; из них 30 индексированы в базе данных OMIM [15]. В первую очередь, мы рассмотрели гены, ассоциированные с известными заболеваниями: PLN, MCM9, TRDN, LAMA2 и GJA1 (таблица).
Гены, индексированные в OMIM и ассоциированные с известными клиническими проявлениями
|
Ген |
Клинические проявления |
Тип наследования |
|
PLN [OMIM: 172405] |
Кардиомиопатия дилатационная, гипертрофическая [OMIM:609909,613874] |
Аутосомно-рецессивный |
|
MCM9 [OMIM: 610098] |
Дисгенезия яичников [OMIM:616185] |
Аутосомно-рецессивный |
|
TRDN [OMIM: 603283] |
Желудочковая тахикардия [OMIM:615441] |
Аутосомно-рецессивный |
|
LAMA2 [OMIM: 156225] |
Врожденная мышечная дистрофия [OMIM:607855] |
Аутосомно-рецессивный |
|
GJA1 [OMIM: 121014] |
1. Дефекты межпредсердной и межжелудочковой перегородок [OMIM:600309] |
Аутосомно-доминантный |
|
2. Краниометафизарная дисплазия [OMIM:218400] |
Аутосомно-рецессивный |
|
|
3. Глазо-зубо-пальцевой синдром [OMIM:164200,257850] |
Аутосомно-доминантный и аутосомно-рецессивный |
|
|
4. Эритрокератодермия [OMIM:133200] |
Аутосомно-доминантный и аутосомно-рецессивный |
|
|
5. Синдактилия, 3 типа [OMIM:186100] |
Аутосомно-доминантный |
Согласно базе данных генетических заболеваний - OMIM, нарушения генов PLN [OMIM:172405] и TRDN [OMIM:603283] приводят к сердечной патологии с рецессивным типом наследования. Сердечной патологии у ребенка выявлено не было. Однако нельзя исключить проявление заболеваний сердца в более позднем возрасте. Нарушения гена MCM9 [OMIM;610098] приводят к дисгенезии яичников с аутосомно-рецессивным типом наследования. Возраст девочки не позволяет рассматривать ассоциацию делеции этого гена с соответствующими его мутации фенотипическими проявлениями. Данные об ассоциации генов PLN, TRDN и MCM9 с нервной и психической патологией отсутствуют.
Учитывая фенотип ребенка, интерес представляют 2 других гена, индексированных в OMIM и локализованных в области делеции, это гены: LAMA2 и GJA1. Нарушения гена LAMA2 [OMIM:156225] вызывают врожденную мышечную дистрофию. Заболевание имеет рецессивный тип наследования и проявляется в виде тяжелой врожденной мышечной дистрофии. Помимо мышечной слабости, характерным изменением является расширение желудочков мозга. Иногда наблюдается умственная отсталость и судороги. У обследуемого ребенка отмечаются мышечная гипотония и задержка психоречевого развития. Исследование с помощью МРТ выявило вентрикуломегалию. Учитывая аутосомно-рецессивный тип наследования, делеция одной копии гена LAMA2 не должна приводить к развитию патологического фенотипа ребенка. Однако такую возможность нельзя исключить в случае, если имеющаяся копия гена содержит нарушения.
Согласно базе данных ОMIM, нарушения гена GJA1 [OMIM:121014] вызывают глазо-зубо-пальцевой синдром [OMIM:164200], известный также как окуло-денто-дигитальная дисплазия. Это генетическое заболевание характеризуется высокой пенетрантностью и широкой фенотипической вариабельностью.
В работе Paznekas с соавторами [16] было описано более 240 пациентов с глазо-зубо-пальцевым синдромом. Типичными черепно-лицевыми аномалиями, характерными для данного синдрома, являются микроцефалия, тонкие гипопластические крылья носа, выраженная носовая перегородка. К другим скелетным аномалиям при данном синдроме относятся черепной гиперостоз, челюсть с высоким альвеолярным гребнем, ретрогнатия верхней и нижней челюстей и широкие трубчатые кости. В большинстве случаев у больных наблюдаются такие аномалии строения зубов, как микродонтия, частичная анодонтия, гипоплазия эмали, множественный кариес и ранняя потеря зубов, а также хрупкие ногти и аномалии волос (гипотрихоз, медленный рост) [17-18]. Микроаномалии конечностей при глазо-зубо-пальцевом синдроме включают: синдактилию третьего, четвертого и пятого пальцев рук, и второго, третьего, четвертого пальцев ног, камптодактилию, клинодактилию, возникающую из-за гипоплазии или аплазии средних фаланг пальцев. К офтальмологическим нарушениям относятся: недоразвитая роговица и конъюнктива, мелкопористый хрусталик, катаракта, глаукома и атрофия зрительного нерва. В некоторых случаях отмечаются диспластичные ушные раковины и нарушения слуховой проводимости. К неврологическим проявлениям глазо-зубо-пальцевого синдрома относятся дизартрия, неврологические расстройства мочеиспускания, судорожные параличи, атаксия, слабость передних большеберцовых мышц и судороги [19]. Симптомы спастического мочевого пузыря или нарушения походки являются обычными неврологическими проявлениями, особенно заметными к возрасту 20 лет. На МРТ головного мозга больных, как правило, определяются диффузные билатеральные аномалии подкоркового белого вещества, которые можно определить, как результат медленно прогрессирующей лейкодистрофии.
Из вышеперечисленных симптомов в литературных источниках у представленного ребенка присутствуют следующие клинические проявления: ретрогнатия, мелкие дистрофичные зубы, тонкие ногти, тонкие редкие волосы, диспластичные ушные раковины, брахидактилия, эпилептические приступы. Со стороны зрения характерных проявлений нет, но была выявлена ангиопатия сетчатки, которая может впоследствии приводить к катаракте и глаукоме. На МРТ обнаружены изменения в виде дилатации желудочков, которые могут быть признаком лейкодистрофии.
Ген GJA1 кодирует белок коннексин 43. Коннексины являются субъединицами межклеточных контактов, которые непосредственно связывают цитоплазму соседних клеток и осуществляют транспорт небольших ионов и сигнальных молекул (<1 кДа), затрагивая все системы организма. Межклеточные коммуникации играют значимую роль в дифференцировке клеток и регуляции роста. Разнообразие белков семейства коннексинов обеспечивает разнообразие межклеточных контактов и их свойств. Ген GJA1 имеет повышенную экспрессию в стволовых клетках эмбриона, плюрипотентных стволовых клетках, гладкой мускулатуре, кератиноцитах и остеобластах. В ЦНС коннексин 43 является важным белком астроцитов и эпендимоцитов, но не экспрессируется на олигодендроцитах и нейронах. По данным литературы, делеция одной копии гена коннексина не приводит к развитию патологического фенотипа [20-21]. Потеря двух аллелей гена GJA1, напротив, имеет негативные последствия, и другие гены семейства коннексинов не в силах компенсировать потерю. У описываемого ребёнка из-за делеции имеется только одна копия гена GJA1, поскольку в фенотипе ребенка присутствуют черты окуло-денто-дигитальной дисплазии, можно предположить, что в единственной копии гена имеются нарушения. Помимо генов, ассоциированных с патологией и описанных выше, делеция затрагивает еще 61 ген, вариации которых, возможно, могут вносить вклад в фенотип представленного ребенка.
Первый эпилептический приступ у девочки произошел в возрасте 1 года, а в дальнейшем был поставлен диагноз криптогенной генерализованной эпилепсии. Мы провели анализ литературных данных по ассоциациям нарушений данного участка и эпилепсии. Действительно, в нескольких работах описана ассоциация интерстициальной делеции хромосомы 6 (6q22q24) и эпилепсии с фебрильно-провоцируемыми приступами при доминантном типе наследования с высокой пенетрантностью и ранней манифестацией. В дальнейших исследованиях участок, отвечающий за различные эпилептические проявления, был сужен до 7,6 млн пн с геномной локализацией в 6q22.1q22.31 (насчитывает 18 генов, индексированных в OMIM). Кроме того, можно предположить, что этот участок может включать новые локусы предрасположенности к идиопатической генерализованной эпилепсии.
В другой работе авторы описали 6 неродственных пациентов с разными делециями внутри участка 6q21q22.31 и установили, что его потеря приводит к задержке психического и речевого развития, ухудшению когнитивных функций, эпилепсии и треморам различной интенсивности [22]. Приступы судорог наблюдались у 3 из 6 пациентов, что совпадает с другими опубликованными данными о возможной неполной пенетрантности генов предрасположенности к эпилепсии при делеции 6q22 [23-24]. Далее исследователи сузили область анализа хромосомы до сегмента величиной 250000 пн в участке 6q22.1. В этой области локализован ген NUS1. Анализ транскрипции генов показал, что NUS1 экспрессируется в мозге. NUS1 кодирует субъединицу мембранного рецептора NgBR, который взаимодействует с регуляторами роста и подвижности нейронов [25]. Исходя из приведенных данных, эпилептические проявления у ребенка могут быть связаны непосредственно с геном NUS1.
Анализ экспрессии генов
Одним из основных параметров при приоритизации генов-кандидатов, нарушения в которых могут приводить к развитию нервных и психических заболеваний, является экспрессия генов в соответствующих тканях. Ранжирование и «фильтрация» генов проводится по уровню их экспрессии в клетках различных тканей. В ходе оценки данных экспрессии 66 генов, входящих в делетированный участок, путём анализа базы данных BiоGРS [26] было установлено, что повышенную экспрессию в различных областях головного мозга имеют 10 генов: GJA1, AKAP7, EPB41L2, FABP7, FAM184A, HDDC2, NKAIN2, PTPRK, SERINC1 и TPD52L1 (рис. 3). Из них гены GJA1, RNF146, SERINC1, TPD52L1 имеют повышенную экспрессию в префронтальной области коры головного мозга.
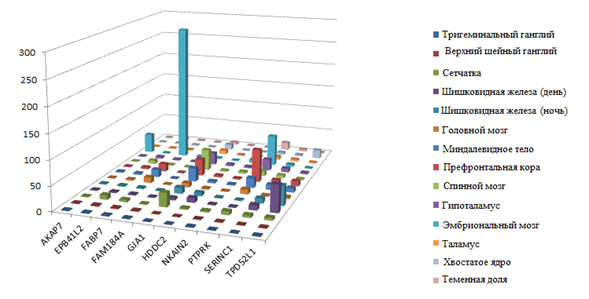
Рис. 3. Экспрессия генов в различных областях мозга (по оси абсцисс гены AKAP7, EPB41L2, FABP7, FAM184A, GJA1, HDDC2, NKAIN2, PTPRK, SERINC1, TPD52L1, уровень экспрессии генов по оси ординат в баллах)
Гены AKAP7, FABP7, NKAIN2, PTPRK характеризуются высокой экспрессией при формировании мозга эмбриона. Нарушение их функции может сказываться на дифференцировке нейронов, что в свою очередь может приводить к общему снижению когнитивной функции и к умственной отсталости. В литературе, однако, информация о нарушениях генов AKAP7, FABP7, NKAIN2, PTPRK отсутствует.
Интерактомный анализ
Был проведён интерактомный анализ (изучение межбелковых взаимодействий) продуктов транскрипции генов, находящихся в области делеции. На рисунке 4 представлена схема взаимодействий белков, кодируемых генами, входящими в область делеции. Схема была смоделирована с использованием онлайн-ресурса [13]. В ходе анализа были выделены две интерактомные сети, представляющие интерес для дальнейшей «фильтрации» процессов-кандидатов (рис. 4).
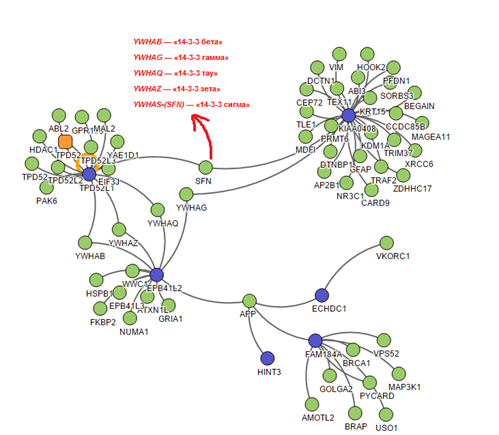
Рис. 4. Схематическое изображение взаимодействий между белками, ассоциированными с генами, локализованными в области делеции (обозначены синим цветом), и другими белками клетки (обозначены зеленым цветом)
Белки EPB41L2, TPD52L1, имеющие повышенную экспрессию в клетках головного мозга, и белок KIAA0408 взаимодействуют с 5 белками (YWHAQ, YWHAZ, YWHAB, YWHAG, SNF), которые относятся к одному классу регуляторных белков семейства 14-3-3[27-28]. Они распознают и специфически взаимодействуют с фосфорилированными аминокислотными остатками белков. Фосфорилирование остатков серина, треонина и тирозина, катализируемое протеинкиназами, является одним из широко распространённых видов посттрансляционных модификаций, влияющих на структуру и свойства белка. Эти белки задействованы в целом ряде значимых биологических процессов, включая регуляцию клеточного цикла, миграцию нейронов, апоптоз, нейрогенез, аксональное наведение и другие процессы в ЦНС. Нарушения этих процессов связывают с патогенезом нервных и психических, а также нейродегенеративных заболеваний [29]. Множество исследований свидетельствуют о связи качественных и количественных нарушений белков семейства 14-3-3 с различными нервными и психическими нарушениями [30].
Другая заслуживающая внимание интерактомная сеть состоит из нескольких белков, кодируемых следующими генами, локализованными в области делеции: EPB41L1, ECHDC1, HINT3, FAM184A, включая белок APP (предшественник бета-амилоида), с которым все вышеперечисленные белки имеют прямое взаимодействие. Экспериментальные данные свидетельствуют о функциональной роли APP в развитии синапсов [31-32]. Нарушение этого белка связывают с развитием одной из форм старческого слабоумия - болезни Альцгеймера. Нарушения в нескольких генах, участвующих в общих клеточных процессах, могут иметь кумулятивный эффект для функционирования целого каскада процессов, в котором они участвуют, что может иметь негативные последствия для ЦНС и организма в целом. Не исключено, что у данного пациента в дальнейшем могут развиться нейродегенеративные нарушения.
Заключение
Представлен уникальный случай интерстициальной делеции длинного плеча хромосомы 6 (q22.1-q23.2), проведён анализ генов, локализованных в этой области, направленный на приоритизацию генов и процессов-кандидатов, ассоциированных с развитием фенотипических проявлений у ребёнка. Ранжирование и «фильтрация» генов проведены по уровню их экспрессии в клетках различных тканей. Среди этих генов наиболее выраженную патогенетическую роль имеет ген GJA1, проявляющийся в виде глазо-зубо-пальцевого синдрома. Похожие функциональные и морфологические нарушения присутствуют у обследованного ребёнка, что позволяет сделать вывод о ключевой роли гена GJA1 в развитии специфического фенотипа. Присутствующие эпилептические проявления, по-видимому, могут быть вызваны делецией гена NUS1, роль которого в развитии эпилепсии доказана. Результаты интерактомного анализа выявили две интерактомные сети, функционирование которых может быть нарушено в результате делеции. Изменения в этих цепях могут приводить к нарушениям функциональной работы мозга.
Представленный случай демонстрирует необходимость применения молекулярно-цитогенетических методов исследования с дальнейшим биоинформатическим анализом с помощью ранжирования и «фильтрации» генов-кандидатов и процессов-кандидатов, приводящих к фенотипическим проявлениям.
Работа поддержана грантами «Российского фонда фундаментальных исследований» (проекты № 17-04-01366А и №16-54-76016 ЭРА_а) и программой фундаментальных исследований Президиума РАН «Фундаментальные исследования для разработки биологических технологий» (ФИМТ) (проекта № ФИМТ-2014-235).
Библиографическая ссылка
Юров И.Ю., Ворсанова С.Г., Демидова И.А., Васин К.С., Шмитова Н.С., Яблонская М.И., Гордеева М.Л., Юров Ю.Б. МОЛЕКУЛЯРНО-ЦИТОГЕНЕТИЧЕСКИЕ И БИОИНФОРМАТИЧЕСКИЕ ИССЛЕДОВАНИЯ ДЕЛЕЦИИ ХРОМОСОМЫ 6 (6q22.1-q23.2): ВОЗМОЖНОСТИ ИНТЕРАКТОМНОГО АНАЛИЗА // Современные проблемы науки и образования. 2017. № 6. ;URL: https://science-education.ru/en/article/view?id=27349 (дата обращения: 10.07.2026).