


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
MOSAIC TRISOMY OF CHROMOSOME 8: THE APPLICABILITY OF MOLECULAR KARYOTYPING

При трисомии хромосомы 8 преобладают мозаичные формы в отличие от многих аутосомных трисомий, где встречаются регулярные формы заболевания. Регулярная трисомия хромосомы 8, вероятно, несовместима с жизнью плода. По некоторым данным, 0,8% спонтанных потерь беременности связаны с трисомией данной хромосомы [1; 2]. Точно установить минимальную долю аномальных клеток пока не представилось возможным, как и установить корреляцию между выраженностью фенотипических проявлений и соотношением трисомных и нормальных клеток в организме. Более того, клинических различий в регулярных и мозаичных формах заболевания, выявленных на культивированных клетках крови, не обнаружено, что позволяет объединить их в одну группу [4; 10; 11]. Дети рождаются доношенными с нормальной массой тела. При рождении и в дальнейшем они не отстают в росте. Наблюдается умеренная задержка умственного развития. Частый признак данной трисомии - поражение головного мозга, в основном в виде агенезии мозолистого тела. Данный порок также встречается и при других хромосомных нарушениях (трисомия 13 - синдром Патау, моносомии 13q и 18p), но при указанных синдромах он сочетается с другими летальными или сублетальными пороками головного мозга. Дети с пороками мозга при трисомии хромосомы 8 доживают в среднем до 12 лет, но встречаются случаи описания пациентов в возрасте и до 17 лет. Из других аномалий мозга при данном нарушении известна гидроцефалия. Определены специфические признаки для данного синдрома, к которым относятся выпуклый лоб, вывернутая нижняя губа, аплазия надколенника, контрактуры суставов, глубокие борозды между межпальцевыми подушечками, пороки мочевой системы. Наблюдаются также косоглазие, эпикант, высокое нёбо, микрогнатия, деформированные ушные раковины с аномальными мочками, короткая складчатая шея, камптодактилия, длинные пальцы, клинодактилия, сколиоз, аномальные тазобедренные суставы, косолапость, паховые грыжи, пороки сердца и желудочно кишечного тракта [1; 2; 8].
Мозаичные перестройки не всегда попадают в поле зрения при цитогенетическом исследовании (кариотипировании) [2; 3; 6-8; 12; 13]. Это связано с тем, что аномальные клетки, как правило, имеют более низкую пролиферацию относительно клеток с нормальным набором хромосом. Если всё же есть предположение о мозаичной анеуплоидии, целесообразно использовать метод флуоресцентной гибридизации in situ (FISH) на интерфазных ядрах. Благодаря данному методу можно установить уровень мозаичных клеток [3; 6; 8; 12-15]. В настоящее время существуют также и другие современные молекулярно-цитогенетические методы, например молекулярное кариотипирование (array CGH), которое эффективно выявляет аномалии генома [4; 7-9; 14], но пока необоснованно редко используется в исследованиях мозаицизма [5-7; 14; 15].
Целью работы
Исследование геномных аномалий у ребенка с пороками развития и «скрытым» низкопроцентным мозаицизмом с помощью современных молекулярно-генетических методов сканирования генома.
Материалы и методы
В настоящей работе было проведено цитогенетическое и молекулярно-цитогенетическое исследование клеточного материала ребёнка семи лет с множественными пороками развития. Препараты метафазных хромосом получали из лимфоцитов периферической крови, культивируемых in vitro стандартным методом. Цитогенетический анализ проводили на хромосомных препаратах с использованием светового микроскопа при увеличении х1125. Хромосомы идентифицировали при помощи дифференциального окрашивания хромосом по длине (G- и C-методы), которые осуществлялись по общепринятым протоколам [1-3; 12]. Молекулярное цитогенетическое исследование проводилось методом серийной сравнительной геномной гибридизации (array CGH) согласно ранее описанному протоколу с использованием SNP/олигонуклеотидной микроматрицы, разрешение не менее 1 тысячи пн (Affymetrix) [5; 7; 9].
Результаты исследования и обсуждение
Представляемый ребёнок семи лет, который родился от 6 беременности, протекавшей на фоне токсикоза с угрозой прерывания. Преждевременные роды были на 35 неделе. Масса тела при рождении составила 2800 г, рост 49 см, оценка по шкале Апгар 7/7. До года ребёнок развивался с задержкой моторного развития (ходить начала с 1 г 6 месяцев), до 2 лет присутствовала выраженная мышечная гипотония, до 3 лет наблюдалась задержка речевого развития. В первые годы жизни ребёнок получал медикаментозную терапию вследствие железодефицитной анемии. Были случаи потери сознания с последующим тоническим напряжением мышц с ознобообразным дрожанием. В дальнейшем поставлен диагноз криптогенная генерализованная эпилепсия (фибрильно провоцируемые приступы). С 2 лет появилось редкое мочеиспускание большими объёмами. При исследовании урологом было сделано заключение о склерозе шейки мочевого пузыря и нейрогенной дисфункции мочевого пузыря. Через некоторое время после рождения ребёнок был оперирован по поводу коарктации аорты и открытого артериального протока. В возрасте семи лет наблюдались следующие микроаномалии развития: короткая шея, пухлые губы, приоткрытый рот, широкий нос, узкий таз. Ребёнок был принят в общеобразовательную школу, но оставлен на второй год из-за низкой успеваемости. Сейчас находится на домашнем индивидуальном обучении.
В результате проведенного цитогенетического анализа был обнаружен кариотип: 46,XY,9qh. Исходя из клинических признаков и отсутствия аномального кариотипа, было проведено молекулярное кариотипирование. При молекулярно-цитогенетическом исследовании SNP/олигонуклеотидная CGH была выявлена мозаичная форма трисомии хромосомы 8 (рис. 1).
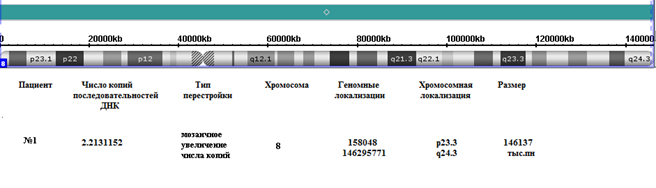
Рис. 1. Схематическое изображение мозаичной трисомии хромосомы 8, обнаруженной с помощью молекулярного кариотипирования.
Учитывая соотношение интенсивности сигналов, можно сделать вывод о том, что трисомия является мозаичной (число аномальных клеток не менее 20%). Следует сказать, что данная хромосома содержит 1484 гена из них в OMIM индексируется 481 ген. Около 8% генов участвуют в развитии и функционировании головного мозга, среди них ген CA8, ассоциированный с умственной отсталостью и церебральной атаксией, ген TMEM67 (синдром Жубер), DDHD2 (спастическая параплегия), RNF170 (аутосомно-доминантная атаксия), POMK (мышечная дистрофия), TAF2 (аутосомно-рецессивная умственная отсталость), RECQL4 (хромосомная нестабильность и ряд наследственных заболеваний).
Исходя из клиники пациента: ЗПРР; склероз шейки мочевого пузыря, нейрогенная дисфункция мочевого пузыря; коарктация аорты и открытый артериальный проток; мышечная гипотония; аномальные тазобедренные суставы; шаркающая походка; короткая шея; широкая спинка носа - можно говорить, что данные клинические проявления схожи с клиникой описанных случаев мозаичной трисомии хромосомы 8. В то же время ряд характерных клинических признаков, таких как поражение головного мозга, аномальные тазобедренные суставы, аплазия надколенников, глубокие борозды между межпальцевыми подушечками, контрактуры различных суставов отсутствовали, что может быть связано с процентным соотношением мозаичных клонов (трисомных и нормальных), которые выявляются в клетках периферической крови. Однако трисомию 8 в других тканях не исследовали. Учитывая этот факт, а также результаты предыдущих молекулярно-цитогенетических исследований [1-3; 6-9; 12-15], следует рекомендовать дополнительные исследования методом FISH на некультивированных клетках других тканей (например, клетках буккального эпителия или фибробластов кожи) для оценки процентного соотношения нормальных и аномальных клонов клеток с целью более детальных корреляций «генотип-фенотип».
Заключение
В заключение необходимо отметить, что в последние годы появляется все больше работ, указывающих на связь низкопроцентного хромосомного мозаицизма с клиническими проявлениями [2; 6-8; 14; 15]. Обнаружение даже незначительной доли клеток с хромосомной аномалией может объяснить причину заболевания, тогда как наличие у больного преобладающей доли нормальных клеток предполагает более благоприятный прогноз течения болезни и менее явное проявление фенотипических признаков [1; 2; 12]. При неявной клинической картине у пациента не всегда удаётся ассоциировать данную патологию с определённым хромосомным нарушением, а при кариотипировании трудно выявить мозаичную форму такой трисомии, как трисомия хромосомы 8, из-за того что клетки с подобными нарушениями в некоторых случаях имеют более низкую пролиферацию по сравнению с нормальными. В последнее время появились такие методы, как молекулярное кариотипирование [2; 3; 7; 9; 15], применение которых считалось неэффективным для выявления низкопроцентного мозаицизма. В литературе представлены случаи, в которых мозаицизм и мозаичные перестройки (до 20% клеток) выявлялись методом молекулярного кариотипирования [5; 7-9]. Настоящий случай подтверждает подобные выводы относительно возможности данного метода.
Исследование выполнено за счет гранта Российского научного фонда (проект № 14-15-00411).
Библиографическая ссылка
Васин К.С., Ворсанова С.Г., Колотий А.Д., Коростелев С.А., Демидова И.А., Юров И.Ю., Гордеева М.Л. МОЗАИЧНАЯ ФОРМА ТРИСОМИИ ХРОМОСОМЫ 8: ВОЗМОЖНОСТИ МЕТОДА МОЛЕКУЛЯРНОГО КАРИОТИПИРОВАНИЯ // Современные проблемы науки и образования. 2015. № 2-1. ;URL: https://science-education.ru/en/article/view?id=17520 (дата обращения: 29.07.2026).