


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
DEVELOPMENT METHOD FOR QUANTITATIVE DETERMINATION TRIAZAVIRIN IN PHARMACEUTICAL COMPOSITIONS FOR NASAL SPRAY USING THE METHOD OF HIGH PERFORMANCE LIQUID CHROMATOGRAPHY

Материалы и методы исследования
В работе использовалась фармацевтическая субстанция триазавирина (ООО "Завод Медсинтез", Россия, соответствующая ФС 000279-141211), стандартный образец фармацевтической субстанции триазавирина (ООО "Завод Медсинтез", Россия, соответствующий требованиям ФС Триазавирин стандартный образец), морская соль («Биотек Марин», Франция: СТП 00480201-363-2012,), вода очищенная.
Количественное определение триазавирина в фармацевтической композиции проводилось с использованием метода ВЭЖХ. Анализ выполнялся на жидкостном хроматографе Flexar F 15 (Перкин Элмер) с УФ детектором. В качестве неподвижной фазы служила хроматографическая колонка «Hydrosphere C18» компании YMC (Япония), 250 мм × 4,6 мм, размер частиц 3 мкм. Подвижная фаза: 10% ацетонитрила – 90% 0,025М водного раствора натрия ацетата тригидрата, рН = 7,40. Скорость потока 0,85 мл/мин. Детектирование осуществляли при длине волны 215 нм. Температура колонки 35±1 0С. Время проведения анализа 20 минут.
Методика. Исследуемый раствор: 2,5 мл исследуемого раствора помещали в мерную колбу вместимостью 25 мл, обрабатывали смесью ацетонитрил - 0,025М водного раствора натрия ацетата тригидрата (10:90), перемешивали в течение 10 минут, доводили до метки подвижной фазой.
Точную навеску стандартного образца триазавирина около 25 мг высушенного до постоянной массы, помещали в мерную колбу вместимостью 25 мл, растворяли в растворе подвижной фазы, перемешивали и доводили до метки растворителем.
Результаты исследования и их обсуждение
Для проведения исследования по количественному определению триазавирина была приготовлена модельная смесь состава: триазавирин 0,2508 г., морская соль 0,2506 г., вода очищенная до 25 мл.
На первом этапе исследования с целью разработки методики количественного определения представляло интерес подобрать оптимальную подвижную фазу (элюент) и условия хроматографирования. При подборе подвижной фазы учитывалась растворимость триазавирина в органических растворителях. Было установлено, что фармацевтическая субстанция триазавирина растворима диметилсульфоксиде, диметилформамиде, ацетонитриле и других органических растворителях. Наличие разнообразных групп (нитрогруппы, азогруппы, метилтиогруппы) в химической структуре триазавирина (рис.1) делает возможным применение метода ВЭЖХ в количественном анализе.
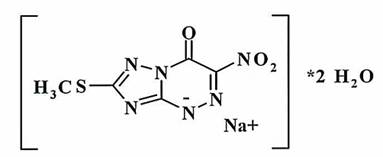
Рис. 1. Химическая структура триазавирина
В результате анализа литературных источников [5] было установлено, что наиболее часто в методе ВЭЖХ в качестве подвижной фазы используется ацетонитрил. Данный элюент находит широкое применение при анализе лекарственных препаратов. Кроме того, ацетонитрил обладает достаточно высокой элюирующей силой, относительно незначительным токсическим действием, низкой вязкостью и доступностью. Было установлено, что триазавирин растворим в ацетонитриле. Также было установлено, что наиболее часто при анализе лекарственных препаратов, являющихся солями сильных оснований, используется сочетание ацетонитрила с 0,025М водным раствором натрия ацетата. Поэтому выбор был сделан в пользу данной подвижной фазы. Следующим этапом был подбор оптимального соотношения ацетонитрила и 0,025М водного раствора натрия ацетата тригидрата. С этой целью были приготовлены растворы, содержащие различное соотношение ацетонитрила и 0,025М водного раствора натрия ацетатата тригидрата (табл. 1).
Таблица 1
Соотношение компонентов в составе подвижной фазы
|
№ подвижной фазы |
Компоненты подвижной фазы |
|
|
Ацетонитрил, % |
0,025М водного раствора натрия ацетата тригидрата, % |
|
|
1 |
5 |
95 |
|
2 |
10 |
90 |
|
3 |
15 |
85 |
|
4 |
20 |
80 |
|
5 |
25 |
75 |
|
6 |
30 |
70 |
Было установлено, что наиболее оптимальное время хроматографирования, значения эффективности и разрешения между пиками триазавирина и его основной примеси 3-метилтио--5-амино-1,2,4-триазола получались при соотношении ацетонитрила и 0,025М водного раствора натрия ацтетата тригидрата (10:90), поэтому был сделан выбор в пользу данного соотношения этих компонентов подвижной фазы.
На следующем этапе была разработана оптимальная методика количественного определения триазавирина в фармацевтической композиции. Методика приготовления исследуемого и стандартного растворов для хроматографирования представлена в разделе «Материалы и методы исследования».
Для анализа 10 мкл испытуемого раствора и 10 мкл раствора стандартного образца субстанции триазавирина попеременно вводили в колонку хроматографа при помощи автосамплера.
На рисунке 2 представлена хроматограмма исследуемого раствора.
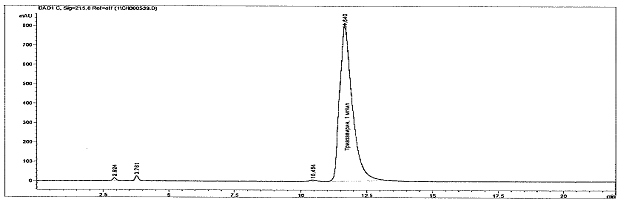
Рис. 2. Хроматограмма исследуемого раствора
Из представленной на рис. 2 хроматограммы видно, что время удерживания триазавирина при анализе исследуемой модельной смеси составило 11,0±0,5 минут, для стандартного образца триазавирина время удерживания составило 11,1±0,5 минут. Время удерживания исследуемого вещества, находящегося в испытуемом растворе, не значительно отличается от времени удерживания стандартного образца триазавирина. Относительное стандартное отклонение времени удерживания не превышает 0,5%.
Содержание триазавирина в мг в 1 мл исследуемого раствора вычисляли по формуле:
Xмг/мл=![]() ,где
,где
S1 – среднее значение площади пика триазавирина на хроматограмме испытуемого раствора, полученное из трех определений;
S0 – среднее значение площади пика триазавирина на хроматограмме раствора стандартного образца, полученное из трех определений;
a0 – навеска стандартного образца субстанции триазавирина, мг;
Р –содержание триазавирина в стандартном образце, %;
2,5; 25; и 25 показатели разведения;
Проверку пригодности хроматографической колонки проводили по следующей методике: 25 мг (точная навеска) СО триазавирина помещали в мерную колбу вместимостью 25 мл., растворяли в растворе подвижной фазы, добавляли 0,25 мл раствора стандартного образца 3 метилтио-5-амино-1,2,4-триазола, перемешивали и доводили до метки растворителем.
Приготовление стандартного образца 3 метилтио-5-амино-1,2,4-триазола: 25 мг (точная навеска) 3 метилтио-5-амино-1,2,4-триазола помещали в мерную колбу вместимостью 25 мл., растворяли в растворе подвижной фазы, доводили до метки тем же растворителем.
С целью проверки пригодности хроматографической системы проводили 5 кратное хроматографирование 10 мкл раствора. В дальнейшем рассчитывали следующие показатели: эффективность колонки, разрешение между пиками, фактор асимметрии. В результате расчетов были получены следующие данные (таблица 2).
Таблица 2
Результаты определения пригодности хроматографической колонки
|
Наименование параметра хроматографической колонки |
Формула расчета |
Полученные значения параметра |
Нормативный показатель для включения в нормативную документацию
|
|
Эффективность колонки для пика триазавирина |
N = 5,54×RT2 / W20,5 , где: RT – время удержания пика; W0,5 – ширина пика на 0,5 высоты |
3524 |
Не менее 3500 теоретических тарелок |
|
Разрешение между пиками триазавирина и 3 метилтио-5-амино-1,2,4-триазола |
Rs = (2×(t’R2 - t’R1))/(W2+W1), где t’R1, t’R2 – приведенные времена удерживания образцовых компонентов*; W1, W2 – ширина пиков образцовых при основании |
5,12 |
Не менее 5 |
|
Фактор ассиметрии триазавирина на высоте 5% от основания |
Т = W0,05/2а, где W0,05 – ширина пика на высоте 5%; a – расстояние от фронта пика до высоты, измеренное на высоте 5% от основания |
1,3 |
Не более 1,5 |
*время удерживания триазавирина составило 11,640 минут, время удерживания 3 метилтио-5-амино-1,2,4-триазола составило 0,46 минут
Результаты, полученные при хроматографировании, позволяют оценить пригодность данной хроматографической системы, а также сделать заключение о том, что данная система может быть использована для анализа триазавирина в составе фармацевтической композиции для назального применения.
Следующим этапом исследования была валидация методики количественного определения триазавирина в фармацевтической композиции.
Валидация представляет собой процесс экспериментального подтверждения того, что методика, используемая для анализа, обеспечивает получение необходимой и достоверной информации об исследуемом веществе, а также может быть пригодна для практического применения [6].
Валидация методики осуществлялась по таким валидационным характеристикам как линейность, прецизионность в условиях повторяемости и базировалась на данных литературных источников [5,6].
Линейность аналитической методики, по данным литературных источников [6], представляет собой наличие линейной зависимости аналитического сигнала (в нашем случае площадь пика триазавирина) от концентрации определяемого вещества в исследуемой пробе в пределах аналитической области методики.
С целью проведения валидации методики по характеристике линейность готовили растворы стандартных веществ из фармацевтической субстанции триазавирина 5 уровней концентрации 10, 50, 75, 100, 125 и 150% от номинального значения с постоянным содержанием в них морской соли (10 мг/мл). Затем проводили разведение, согласно описанной выше методики и хроматографировали полученные пробы. Далее проводили измерение аналитического сигнала, в качестве которого выступала площадь пика для анализируемых проб. Оценку линейности проводили путем расчета коэффициента корреляции (значение должно быть не менее 0,999). Результаты анализа представлены в таблице 3.
Таблица 3
Результаты определения триазавирина в фармацевтической композиции (оценка линейности)
|
Навеска триазавирина, г |
Концентрация раствора, мг/мл |
Площадь пика |
|
0,0256 |
0,0256 |
1820822 |
|
0,1253 |
0,1253 |
9057475 |
|
0,1874 |
0,1874 |
13118000 |
|
0,2510 |
0,2510 |
18114951 |
|
0,3125 |
0,3125 |
21903822 |
|
0,3752 |
0,3752 |
2651700 |
На основании полученных данных была построен график зависимости площади пика от концентрации триазавирина (рис.3).
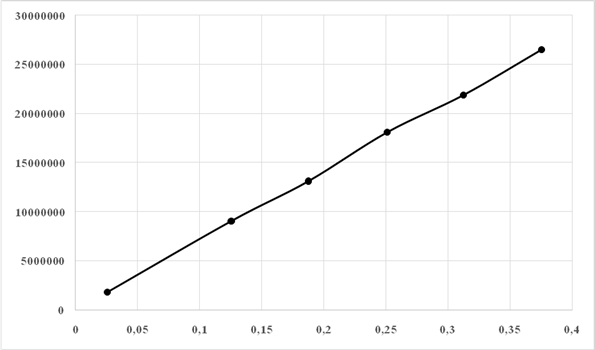
Рис. 3. График зависимости площади пика (по оси ординат) от концентрации триазавирина (по оси абсцисс)
Из рисунка 3 видно, что наблюдается линейная зависимость площади пика от концентрации триазавирина (y =7×107 +100409; r = 0,9995) в диапазоне концентраций от 10 до 150% от номинального значения, т.е. в интервале от 0,02 до 0,4 мг/мл. Коэффициент корреляции равен 0,9995, что соответствует рекомендуемым значениям (не менее 0,999).
На следующем этапе оценивалась прецизионность методики в условиях повторяемости.
Повторяемость – это первый уровень прецизионности. Оценка методики количественного определения проводится по независимым результатам, получаемых в одинаковых условиях в одной лаборатории (одним и тем же аналитиком, на одном и том же оборудовании, с использованием одного и того же набора реактивов) в пределах короткого промежутка времени [6].
Определение показателя прецизионность методики в условиях повторяемости проводили на одном исследуемом образце (состав: триазавирин 0,2508 г., морская соль 0,2506 г., вода очищенная до 25 мл.). Определение было проведено 6 кратно, количественное концентрацию триазавирина в растворе определяли по градуировочному графику с учетом разведения. Затем рассчитывали относительное стандартное отклонение. Результаты определения прецизионности в условиях повторяемости для триазавирина представлены в таблице 4.
Таблица 4
Результаты определения прецизионности в условиях повторяемости для триазавирина
|
№ |
Площадь пика триазавирина |
Найденная концентрация триазавирина, мг/мл |
|
1 |
17792909 |
10,11 |
|
2 |
17704912 |
10,06 |
|
3 |
17968902 |
10,21 |
|
4 |
18114951 |
10,29 |
|
5 |
18440409 |
10,48 |
|
6 |
18580409 |
10,56 |
|
|
- |
10,246 |
|
SD |
- |
0,2003 |
|
RSD,% |
- |
1,95 |
Расчет показал, что относительное стандартное отклонение составило 1,95%. Полученные данные говорят о том, что методика обладает достаточно хорошей прецизионность в условиях повторяемости.
Заключение
Разработанная методика количественного определения триазавирина в фармацевтической композиции для назального применения с использованием метода ВЭЖХ показала хорошие результаты, следовательно, может быть использована в количественном анализе с последующим включением ее в нормативную документацию на ЛФ.
Рецензенты:Русинов В.Л., д.х.н., профессор директор химико-технологического института Уральского федерального университета им. Первого Президента России Б.Н. Ельцина, г.Екатеринбург;
Денисенко О.Н., д.фарм.н., профессор, декан ФУП, заведующий кафедрой фармации Пятигорского МФИ филиала ГБОУ ВПО Волгоградский ГМУ Минздрава России, г. Пятигорск.
Библиографическая ссылка
Кинев М.Ю., Гаврилин М.В., Мельникова О.А., Петров А.Ю. РАЗРАБОТКА МЕТОДИКИ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ТРИАЗАВИРИНА В ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ ДЛЯ НАЗАЛЬНОГО СПРЕЯ С ИСПОЛЬЗОВАНИЕМ МЕТОДА ВЭЖХ // Современные проблемы науки и образования. 2014. № 6. ;URL: https://science-education.ru/en/article/view?id=16499 (дата обращения: 19.07.2026).