


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
MODELING OF PENICYLLINAMIDASE ENANTIOSELECTIVITY BY FREE ENERGY PERTURBATION

Введение
В последнее время большой интерес представляет разработка методов моделирования энантиоселективности мутантных форм ферментов, позволяющих значительно сократить объем экспериментальной работы при создании новых эффективных биокатализаторов с заданными свойствами. Наиболее часто для данных целей применяется метод молекулярного докинга [6], который характеризуется высокой производительностью, но сравнительно низкой точностью, обусловленной применением эмпирических силовых полей и недостаточной степенью сэмплирования степеней свободы системы. Более корректным и, как следствие, точным методом моделирования является метод возмущения свободной энергии (ВСЭ), позволяющий использовать полноатомные модели белка и растворителя и проводить усреднение по большому количеству микросостояний системы. Данный метод основан на определении изменения свободной энергии при небольшом постепенном изменении моделируемой системы (например, замена метильной группы на этильную) в ходе молекулярной динамики. Потенциально он может быть применён и для расчёта энергии перехода энантиомерных соединений друг в друга (и, как следствие, расчёта энантиоселективности). Таким образом, создание нового высокоточного подхода к определению энантиоселективности in silico является востребованной и актуальной задачей.
В качестве объекта исследования была выбрана пеницилламидаза (пенициллинацилаза, ПА) – фермент, который катализирует синтез и гидролиз амидных субстратов, и обладает стереоселективностью [2], что позволяет использовать её не только для синтеза промышленно важных пенициллинов, но и для разделения оптически активных субстратов [7]. ПА экспрессируется в бактерии Escherichia coli, что позволяет достаточно легко вводить в данный фермент те или иные мутации методами молекулярной биологии для улучшения различных каталитических свойств, в том числе стереоселективности, и нарабатывать данный фермент в значительных количествах [3].
В рамках выполнения настоящего исследования была проведена разработка и валидация методики компьютерного моделирования энантиоселективности пенициллинацилазы.
Методы
Подготовка полноатомной модели и топологического файла комплекса фермент ‑ 2-фенил-N-(1-фенетил)ацетамид (ES). Использовалась трёхмерная структура ПА из базы данных PDB (PDB ID 1GM9), трёхмерная структура энантиомеров 2-фенил-N-(1-фенетил)ацетамида подготавливалась с помощью программы ACD LABS 12 [1]. Для подготовки трёхмерной структуры комплекса проводился молекулярный докинг амидного субстрата в активный сайт фермента с помощью программы Lead Finder [9]. Лучшая поза отбиралась по результатам визуализации комплекса на основании наличия характеристичных водородных связей амидного азота субстрата с остатками Ala61 и Asn241 белка.
Для подготовки топологического файла силового поля OPLS-AA использовалась программа ACPYPE [8]. Окончательную трёхмерную структуру и топологический файл подготавливался с помощью программы pdb2gmx пакета Gromacs [10]. Для добавления молекул растворителя (вода TIP4P) использовалась программа genbox пакета Gromacs.
Подготовка полноатомной модели и топологических параметров тетраэдрического интермедиата (рис. 1). Использовалась трёхмерная структура ПА из базы данных PDB (PDB ID 1GM9), трёхмерная структура тетраэдрического интермедиата оптимизировалась с помощью программы ACD LABS 12 [1]. Для подготовки структуры интермедиата проводится ковалентный докинг тетраэдрического интермедиата в активный сайт фермента с ковалентной привязкой атома OG серина 1 β-цепи к «карбонильному» атому кислорода ацильного донора (в данном случае соединенному с атомом азота нуклеофила) с помощью программы Lead Finder [9]. Лучшая поза отбиралась по результатам визуализации комплекса на основании наличия водородных связей с остатками Ala61 и Asn241.
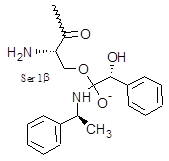
Рисунок 1. Структурная формула тетраэдрического интермедиата
Для подготовки топологического файла силового поля OPLS-AA использовалась программа ACPYPE [8]. Полученные топологические параметры (заряды и типы атомов, а также параметры связей и двугранных углов) вносились в файл aminoacids.rtp, используемый программным пакетом Gromacs [10] для определения параметров аминокислотных остатков. Дополнительно в файл aminoacids.hdb вносились изменения, необходимые для автоматического определения положения атомов водорода.
Окончательная трёхмерная структура и топологический файл тетраэдрического ацилфермент-нуклеофильного комплекса генерировались с помощью программы pdb2gmx пакета Gromacs. Для добавления молекул растворителя (вода TIP4P) использовалась программа genbox пакета Gromacs.
Планаризация энантиомеров комплексов методом ВСЭ. Для определения разницы свободной энергии между двумя различными энантиомерами использовалась схема, отображенная на Рисунке 2.
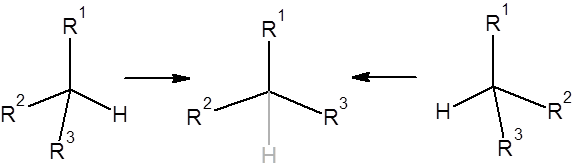
Рисунок 2. Схема расчёта разницы свободной энергии энантиомеров методом ВСЭ.
Для превращения энантиомеров в ахиральное промежуточное состояние, отображенного на Рисунке 2, сначала приводился к нулю заряд на атоме водорода, а затем приводились к нулю Ван-дер-Ваальсовы параметры и валентные и двугранные углов, связанные с данным атомом водорода. Данная процедура проводилась для обоих энантиомеров комплекса белок-субстрат и тетраэдрического интермедиата.
Расчёты разницы свободной энергии энантиомеров методом ВСЭ. Переход методом ВСЭ в каждом случае состоял из 10 равномерных шагов по λ и проводился по следующей схеме (длина шага 2 фс):
- Минимизация структуры до градиента энергии 100 кДж/моль/нм (5000 шагов)
- NVT ‑ динамика (100 пс, 50000 шагов)
- NPT ‑ динамика (100 пс, 50000 шагов)
- Итоговая NPT – динамика (2000 пс, 1000000 шагов)
Изменение свободной энергии рассчитывается при помощи утилиты g_bar:
Результаты и обсуждение
Катализ реакции гидролиза амидного субстрата под действием ПА включает стадию образования нековалентного комплекса ES, ковалентного тетраэдрического интермедиата (лимитирующая стадия для k2), отщепление нуклеофила с образованием ацилфермента и последующий гидролиз ацилфермента [5] (Рисунок 3).
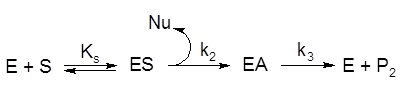
Рисунок 3. Кинетическая схема ПА-катализируемой реакции гидролиза амида. E – свободный фермент, S – амидный субстрат, Nu –нуклеофил (амин), P2 – продукт реакции, образующийся при гидролизе ацилфермента (кислота), ES – фермент-субстратный комплекс, EA – ацилфермент
В случае одного ацильного донора P2, но различных нуклеофилов, энантиоселективность реакции гидролиза будет определяться отношением констант KS и k2. Отношения этих констант для двух энантиомеров могут быть рассчитаны методом возмущения свободной энергии. Итоговая энантиоселективность (для одинаковых начальных концентраций обоих энантиомеров) может быть рассчитана по формуле:
![]()
Согласно разработанной методике, рассчитываются энергии перехода каждого из энантиомеров комплекса фермент-субстрат и тетраэдрического интермедиата в тригональное ахиральное состояние (![]() ,
, ![]() ,
,![]() ,
, ![]() ). На основе полученных значений отношение констант равновесия образования нековалентного комплекса
). На основе полученных значений отношение констант равновесия образования нековалентного комплекса ![]() и
и ![]() может быть определено согласно следующей формуле:
может быть определено согласно следующей формуле:
![]() , поскольку энергии энантиомеров в растворе одинаковы.
, поскольку энергии энантиомеров в растворе одинаковы.
Отношение констант скорости ![]() , которое и характеризует стереоселективность реакции в условии насыщения субстратом, может быть оценено исходя из значения свободной энергии переходов для нековалентного комплекса и тетраэдрического интермедиата по следующей формуле:
, которое и характеризует стереоселективность реакции в условии насыщения субстратом, может быть оценено исходя из значения свободной энергии переходов для нековалентного комплекса и тетраэдрического интермедиата по следующей формуле:
![]()
Анализ молекулярно-динамических траекторий показал, что предложенный способ позволяет достичь перехода от D- и L- энантиомеров к планарному состоянию, как в случае исходного фермент-субстратного комплекса, так и в случае тетраэдрического интермедиата (рисунок 4). Использование протокола ВСЭ при этом позволило определить точное изменение свободной энергии в этих переходах, которое в свою очередь используется для расчёта энантиоселективности фермента.
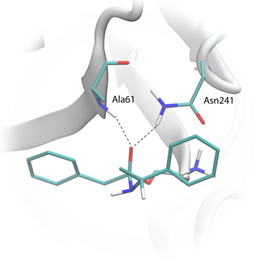
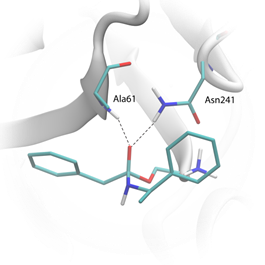
Рисунок 4. Структура тетраэдрического интермедиата с 2-фенил-N-(1-фенетил)ацетамидом: (D)-энантиомер (слева), планарное состояние (справа).
Подстановка результатов расчётов (таблица 1) в формулу для расчёта соотношения ![]() приводит к энантиоселективности 1,79-2,88 с преобладанием L-изомера (разница в свободной энергии активации реакций в 2000±590 Дж/моль). Полученное направление селективности соответствует литературным данным, и достаточно хорошо количественно согласуется с ними (эксп. L/D = 10) [4].
приводит к энантиоселективности 1,79-2,88 с преобладанием L-изомера (разница в свободной энергии активации реакций в 2000±590 Дж/моль). Полученное направление селективности соответствует литературным данным, и достаточно хорошо количественно согласуется с ними (эксп. L/D = 10) [4].
Таблица 1. Итоговые значения свободных энергий тригонализации для гидролиза 2-фенил-N-(1-фенетил)ацетамида, кДж/моль.
|
1 |
|
|
|
|
|
D |
-9,57±0,04 |
-25,84±0,28 |
-9,20± 0,23 |
-14,32±0,14 |
|
L |
-8,75±0,20 |
-28,63±0,32 |
-8,59± 0,10 |
-15,02±0,20 |
Выводы
Предложен новый высокоточный подход к моделированию энантиоселективности ферментативных реакций, основанный на расчёте изменения свободной энергии активации энантиомеров субстрата с помощью метода возмущения свободной энергии. Данный подход позволил явным образом учесть не только атомы реакционного центра, но и остальные атомы белка, а также молекулы растворителя, а также провести усреднение полученных значений энергии по термодинамическому ансамблю состояний системы. Применение подхода к расчёту энантиоселективности гидролиза 2-фенил-N-(1-фенетил)ацетамида пенициллинацилазой показало корректность предложенной схемы перехода, т.к. D- и L-энантиомеры субстрата и тетраэдрического интермедиата переходили в идентичное плоское состояние. Сопоставление полученных результатов с экспериментальными данными показало, что предсказанное направление энантиоселективности (L>D) является правильным, а степень его проявление удовлетворительно согласуется с экспериментом (2,3 – расчётное соотношение L/D, 10 – экспериментальное). В сочетании с методами моделирования структур мутантных ферментов данный подход может быть успешно использован для дизайна новых эффективных стереоспецифичных биокатализаторов.
Работа была поддержана грантом Министерство образования и науки Российской Федерации (соглашение № 14.U02.21.1913 от 04 октября 2012 г.).
Рецезенты:
Швядас В.-Ю.К., д.х.н., профессора факультета биоинженерии и биоинформатики Московского государственного университета имени М.В.Ломоносова, г.Москва.
Анаников В.П., д.х.н., зав. лаб. металлокомплексных и наноразмерных катализаторов Института органической химии им. Н.Д. Зелинского РАН, г.Москва.
Библиографическая ссылка
Зейфман А.А., Перепёлкин В.В., Новиков Ф.Н., Строганов О.В., Чилов Г.Г. МОДЕЛИРОВАНИЕ СТЕРЕОСЕЛЕКТИВНОСТИ ПЕНИЦИЛЛИНАМИДАЗЫ МЕТОДОМ ВОЗМУЩЕНИЯ СВОБОДНОЙ ЭНЕРГИИ // Современные проблемы науки и образования. 2013. № 5. ;URL: https://science-education.ru/en/article/view?id=10601 (дата обращения: 07.07.2026).