



Введение
Структурные несбалансированные хромосомные перестройки относятся к редким наследственным и врожденным заболеваниям. Практически все хромосомные болезни характеризуются тяжелыми клиническими проявлениями, включающими задержку умственного и физического развития, а также сложным комплексом пороков развития. «Классическое кариотипирование» с применением методов дифференциального окрашивания хромосом при постановке диагноза этим больным в большинстве случаев необходимо дополнить современными молекулярно-цитогенетическими методами, такими как флуоресцентная гибридизация in situ (FISH) [1; 2; 4] или сравнительная геномная гибридизация на ДНК-биочипах (array CGH) [5].
В настоящее время известно несколько десятков случаев трисомии 7q, возникающих в результате дупликации сегмента 7q11- (q22,q31,q32)-7qter. Большинство наблюдений связано с унаследованными транслокационными перестройками, реже инсерциями или инверсиями [17; 25; 30; 32; 41]. Клиническая картина заболевания зависит от фрагментов длинного плеча хромосомы 7 (7q), вовлечённых в перестройку. Частичная трисомия участка 7q2àqter коррелирует с нарушением психомоторного развития, аномалиями лица с узкими глазными щелями, эпикантом, массивной нижней челюстью, общей гипотонией. Вариабельность клинических проявлений при этой редкой хромосомной аномалии не позволяет выделить частичную трисомию участка 7q2àqter в отдельный синдром. При частичной трисомии с вовлечением участка 7q3àqter отмечают пренатальную гипоплазию, задержку психомоторного развития, черепно-лицевые аномалии (круглое лицо, высокий, выступающий лоб, эпикант, короткий нос, расщелину нёба, деформированные низкорасположенные ушные раковины) [2; 3]. Также встречаются косоглазие, колобома радужки, большой язык, микроретрогнатия, поперечная ладонная складка, деформация позвоночника и конечностей, низкий рост, отставание речевого развития [25; 41]. В ранних работах частичную трисомию 7q3àqter выделяли в отдельный синдром [15]. Однако накопление подобных случаев и отсутствие явной корреляции фенотип-генотип при частичной трисомии 7q3àqter ставит под сомнение правомочность выделения этой аномалии кариотипа в отдельный хромосомный синдром.
Целью настоящей работы является подробное клиническое и цитогенетическое описание редкого варианта частичной трисомии 7q с ранее не описанной локализацией точек разрыва в участках 7q22-q32. Для установления точного цитогенетического диагноза у больного ребенка нами было использовано сочетание методов классического кариотипирования с использованием G-окрашивания и молекулярно-цитогенетичекого анализа (FISH-метода), что позволило выявить редкую несбалансированную хромосомную аномалию у пробанда с верификацией точек разрыва и определить наличие сбалансированной хромосомной перестройки у матери пробанда. Проведен анализ описанных ранее случаев частичной трисомии хромосомы 7 и обсуждается возможность клинико-генетической корреляции и медико-генетического консультирования в случаях редких хромосомных болезней, требующих обязательного применения современных диагностических молекулярно-цитогенетических технологий.
Материал и методы
На цитогенетическое исследование поступила девочка (1 год 4 месяца) с задержкой психомоторного и речевого развития. Ребёнок от 5-й беременности, протекавшей с угрозой прерывания во всех триместрах, 3-их родов. Родители фенотипически здоровы. Брак неродственный. Первая беременность закончилась рождением дочери с аномалиями развития (кариотипирование не проводилось). Вторая и третья беременности завершились самопроизвольными абортами на разных сроках, от четвёртой родилась девочка с врождённым пороком сердца и грубой задержкой психомоторного развития. Умерла от бронхопневмонии в возрасте 1 года (рис. 1).
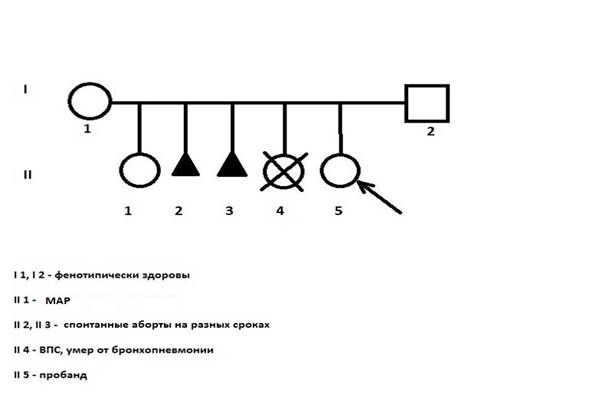
Рис. 1. Родословная семьи пробанда.
Пробанд родилась на 39 неделе, самопроизвольными родами. Масса тела при рождении 3250 г (< 75 центилей), длина 58 см (> 97 центилей). Оценка по шкале Апгар 7/8. Девочка дисгармоничного телосложения с неравномерным распределением по телу жировой клетчатки. Наблюдалась деформация головы с уплощённым затылком, брахицефалия, антимонголоидный разрез глаз, эпикант, длинные ресницы, низко посаженные оттопыренные ушные раковины, выступающая верхняя челюсть, макроглоссия, уплощённая грудная клетка и стопы, арахнодактилия, нарушение дерматоглифики. На момент осмотра окружность головы пробанда была 41 см. Ребёнок не гулил, не сидел, на ноги не опирался, голову не держал, имел отсутствующий взгляд. По заключениям специалистов, у пробанда присутствует тугоухость, частичная атрофия зрительных нервов обоих глаз, гипоксически-ишемическая энцефалопатия с участками кистозно-глиозной аплазии коры теменно-затылочной области, открытая смешанная компенсированная гидроцефалия, замедление миеленизации головного мозга, атрофические изменения лобно-височных отделов, вторичная вентрикуломегалия, гипоплазия мозолистого тела атрофические изменения мозжечка.
Результаты
Цитогенетическая диагностика
Нами было проведено кариотипирование стандартным методом на метафазах с использованием G-окраски с разрешением до 300-500 дискретных G-сегментов на кариотип. Цитогенетический анализ выявил дополнительный генетический материал в длинном плече хромосомы 1 неизвестного происхождения (рис. 2).
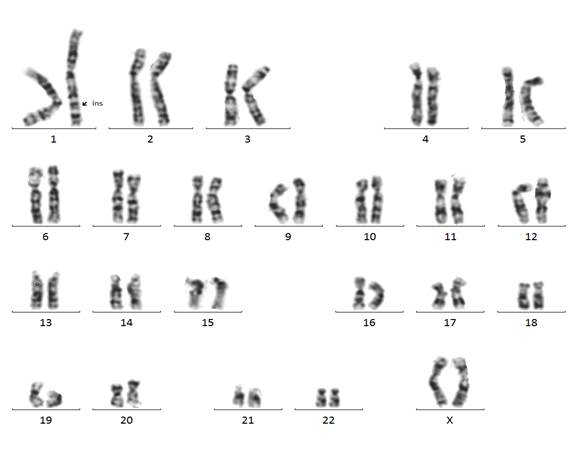
Рис. 2. Кариотип пробанда (G-окраска). В длинном плече хромосомы 1 обнаружен дополнительный генный материал неизвестного происхождения.
При подобных структурных аномалиях необходимо проведение цитогенетического обследования родителей, что позволяет определить, являются ли родители носителями сбалансированной хромосомной перестройки, или же аномалия у ребенка возникла de novo. В результате кариотипирования у матери ребёнка обнаружена производная хромосомы 7 с перицентрической инверсией и отсутствием части генетического материала, который, по-видимому, был перенесен в хромосому 1. Следовательно, у матери наблюдалась сбалансированная хромосомная перестройка, не имеющая клинического проявления. Кариотип пробанда (больной девочки) был определен как 46,XX,der(1) ins(1;7), т.е. фрагмент хромосомы 7 представлен в трех копиях: две в морфологически нормальных хромосомах 7-й пары и одна - в деривативной хромосоме 1. Таким образом, ребёнок имеет несбалансированную хромосомную аномалию в виде частичной трисомии по хромосоме 7. Однако остался невыясненным вопрос о конкретном фрагменте этой хромосомы, перешедшем в хромосому 1, поскольку при G-окраске участки 7q21 и 7q31 идентичны и определение точек разрыва в хромосоме 7 при дифференциальном G-окрашивании невозможно. Этот вывод подтверждается литературными данными. В частности, ранее было описано два клинически различных случая с предполагаемой идентичной трисомией 7q, причём указывается на возможность ошибки трактовки удвоенного сегмента, т. к. различить dup (7)(q11-q22) и dup(7)(q22-q32) при G-окраске невозможно [32].
Молекулярно-цитогенетический анализ
Для уточнения точек разрыва при данной структурной перестройки нами было проведено молекулярно-цитогенетическое исследование (FISH диагностика) с ДНК-зондом MCG-P-405-01 на участок хромосомы 7q21.3 из коллекции сайт-специфических ДНК-зондов лаборатории цитогенетики и геномики психических заболеваний НЦПЗ РАМН [1; 3; 5; 39; 43]. Фрагмент 7q21.3 остался в перестроенной хромосоме 7 (рис. 3). Анализ результатов молекулярного анализа и G-окрашивания свидетельствует о том, что в хромосому 1 «перешёл» участок 7q21, расположенный проксимальнее участка 7q21.3. По данным FISH-диагностики, участок 7 q21.3 остался в перестроенной хромосоме 7, а в хромосому 1 переместился фрагмент 7q31 (рис. 3). Таким образом, описанная перестройка имеет четыре точки разрыва в хромосоме 7: в локусах при инверсии: p11.2,q11.23 и при инсерции 7q22 -q32 в хромосому 1. Следовательно, кариотип матери можно записать как 46,XX,ins(1;7)(q32;q22q32),inv(7)(p11.2;q11.23). С учётом этих данных кариотип дочери верифицируется как 46,XX, der(1) ins(1;7)(q32;q22q32). Таким образом, у больного ребёнка выявлена частичная трисомия по длинному плечу хромосомы 7 в сегменте 7 q22-q32.
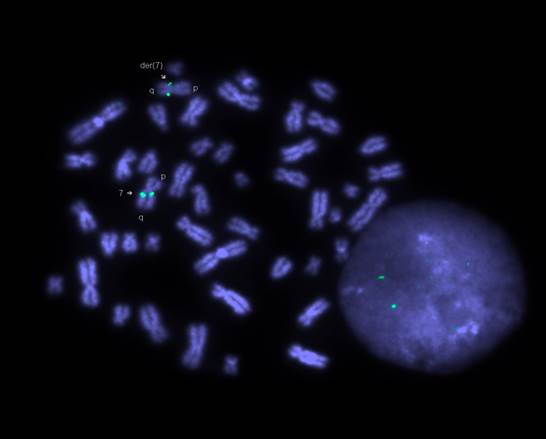
Рис. 3. FISH-диагностика (кариотип матери). Сигнал сайт-специфичного ДНК-зонда (зеленые точки) на участок хромосомы7q21.3 имеется в нормальной хромосоме 7 и остаётся в длинном плече der(7).
Обсуждение
В литературе описано около 50 случаев частичной трисомии длинного плеча хромосомы 7 [25; 30; 32; 41]. Все они касаются дупликации разных сегментов и отличаются вариабельностью описываемых клинических признаков. Многими авторами были предприняты попытки систематизации данных хромосомных аберраций и поиска корреляций фенотип-генотип. Например, было высказано предположение о наличии двух разных хромосомных синдромов с частичными дупликациями хромосомы 7q [25]. Первый предполагаемый синдром включал дупликацию дистального сегмента 7q31-qter с клиническими проявлениями в виде низкого веса и роста при рождении, аномалий черепа, отсутствия микроцефалии, а также характеризовался низкопосаженными ушными раковинами, аномалиями мышечного тонуса и умственной отсталостью. Второй синдром был ассоциирован с трисомией 7q21(22)- q31 и фенотипическими проявлениями в виде узких глазных щелей, эпиканта, уплощённого переносья, задержкой роста, гипотонией. В то же время Novales et al. [25] при анализе литературных данных условно разделили всех пациентов с частичной трисомией по длинному плечу хромосомы 7 на три группы. Для пациентов с дупликацией 7q21(22)- q31, отнесённых к группе 1, были характерны определённые лицевые аномалии (узкие глазные щели, эпикант, страбизм, гипертелоризм, лобная борозда), умственная отсталость и гипотония. Пациенты 2-й группы с дупликацией 7q31-qter имели низкий вес при рождении и лицевые аномалии (узкие глазные щели, гипертелоризм, маленький нос, низкопосаженные деформированные ушные раковины, микроретрогнатию, расщелину нёба), умственную отсталость, скелетные аномалии, раннюю смертность. Третью группу составили пациенты с трисомией по локусу 7q32-qter, с низким весом при рождении, лицевыми дисморфиями (низкопосаженные ушные раковины, маленький нос), скелетными аномалиями, гипотонией, умственной отсталостью. Позже Courtens et al. (2001) описали четвёртую группу пациентов, имеющих трисомию по фрагменту 7q21(22)-qter [14]. Фенотип данных пациентов характеризовался лицевыми аномалиями (высокий лоб, гипертелоризм, микрогнатия, низкопосаженные деформированные ушные раковины, расщелина нёба), короткой шеей, гипотонией, скелетными аномалиями и ранней смертью. Однако частичные трисомии ограничиваются не только перечисленными выше группами. Одни авторы описали клиническую характеристику трисомии по проксимальным районам длинного плеча хромосомы 7, а другие - по более дистальным [8; 18; 21; 28; 37].
В табл. 1 суммированы клинические описания 36 пациентов, полученные разными авторами. Мы постарались представить все случаи частичной трисомии 7q, известные к настоящему времени, они систематизированы по сегментам, участвующим в трисомии 7q. Анализ этих данных не обнаружил серьёзных различий между вариантами трисомий длинного плеча хромосомы 7. Задержка умственного развития характерна для всех случаев трисомии 7q, так же как низкий вес при рождении, наличие передней борозды и низкопосаженные ушные раковины. Одновременно с этим гипотония, которая характерна для трисомии q22-qter и q33-qter, встречается при удвоениях фрагментов q32-qter и q22-q31, но отсутствует при q31-qter. Ранняя смерть (в первый год жизни) была выявлена у пациентов с фрагментами q22-qter и q31-qter, что не было выявлено у больных с вовлечением более дистальных фрагментов (табл. 1). У девочки, описанной в настоящей работе, часть клинических признаков совпала с фенотипическими проявлениями при других трисомиях длинного плеча хромосомы 7. Прежде всего, это касается умственной отсталости. Однако этот признак характерен для большинства хромосомных перестроек. С разной степенью частоты реже встречаются деформация головы, аномалии скелета, эпикант, низкорасположенные деформированные ушные раковины, длинные ресницы, макроглоссия, задержка психомоторного и речевого развития, гипотония (табл. 1).
Таблица 1. Клинические особенности частичной трисомии 7q.
|
Число случаев |
Сегмент
|
Низкий вес при рожде- нии |
Макро-цефалия |
Эпикант |
Низкопо-саженные ушные раковины |
Страбизм |
Маленький нос |
Макро-глоссия |
Передняя борозда |
Скелетные аномалии |
Гипото-ния |
Ранняя смерть |
Умствен- ная отста-лость |
Авторы |
|
|
1 |
q11.22-q11.23 |
+ |
+ |
- |
+ |
- |
- |
|
- |
+ |
- |
+ |
- |
Hoo [18] |
|
|
2 |
q11-q22 |
- |
+ |
- |
+ |
- |
- |
|
+ |
- |
- |
- |
+ |
Kardon [21] |
|
|
3 |
q11-qter |
+ |
- |
+ |
+ |
+ |
- |
|
+ |
+ |
- |
- |
+ |
Wahrman [40] |
|
|
4 |
q21.2-22.1 |
+ |
+ |
- |
+ |
+ |
- |
|
+ |
+ |
+ |
- |
+ |
Lukusa[22] |
|
|
5 |
q21.12-q31 |
+ |
- |
- |
+ |
- |
+ |
|
- |
- |
- |
- |
- |
Humphrey [19] |
|
|
6 |
q21.2-q31.31 |
+ |
+ |
+ |
|
+ |
|
|
+ |
|
+ |
- |
+ |
Weimer [41] |
|
|
7 |
q22-q31 |
- |
|
+ |
+ |
+ |
+ |
- |
- |
- |
- |
- |
+ |
Grace [17] |
|
|
8 |
q22-q31 |
- |
|
+ |
- |
+ |
+ |
- |
+ |
- |
+ |
- |
+ |
Berger [10] |
|
|
9 |
q22-q31 |
+ |
|
- |
+ |
+ |
- |
- |
+ |
- |
+ |
- |
+ |
Serville [29] |
|
|
10 |
q22-q31 |
+ |
- |
- |
- |
+ |
+ |
- |
+ |
|
+ |
- |
+ |
Megarbane[26] |
|
|
11 |
q22-q31.2 |
+ |
+ |
+ |
+ |
+ |
+ |
- |
+ |
+ |
- |
- |
+ |
Romain [28] |
|
|
12 |
q22-q32 |
- |
- |
+ |
+ |
- |
|
+ |
|
+ |
+ |
- |
+ |
Настоящий случай |
|
|
13 |
q22-q34 |
+ |
- |
|
|
+ |
+ |
|
+ |
- |
|
|
+ |
Stratton [35] |
|
|
14 |
q22-qter |
+ |
+ |
- |
- |
- |
- |
|
- |
+ |
+ |
+ |
+ |
Rodriguez [27] |
|
|
15 |
q22-qter |
+ |
|
- |
+ |
- |
- |
+ |
+ |
- |
+ |
+ |
|
Carpenter [13] |
|
|
16 |
q31-qter |
+ |
|
|
+ |
- |
+ |
|
- |
+ |
|
|
- |
Alfi [6] |
|
|
17 |
q31-qter |
+ |
- |
+ |
- |
+ |
+ |
Vogel [38] |
|||||||
|
18 |
q31-qter |
+ |
|
+ |
- |
+ |
|
- |
|
+ |
|
Al Saadi [7] |
|||
|
19 |
q31-qter |
- |
|
- |
+ |
+ |
- |
|
+ |
+ |
|
+ |
+ |
Berger [ 11] |
|
|
20 |
q31.2-qter |
- |
+ |
- |
+ |
- |
- |
|
+ |
+ |
- |
- |
+ |
Rodriguez [27] |
|
|
21 |
q32-qter |
+ |
- |
- |
- |
+ |
- |
+ |
- |
- |
+ |
Newton [24] |
|||
|
22 |
q32-qter |
+ |
- |
+ |
- |
- |
+ |
+ |
- |
- |
+ |
Bass [9] |
|||
|
23 |
q32-qter |
-/+ |
+/- |
+/- |
-/- |
+/+ |
-/+ |
-/- |
+/ |
+/- |
-/- |
+/+ |
Moric-Petrovic* [23] |
||
|
24 |
q32-qter |
-/+ |
|
+/ |
-/+ |
-/ |
-/+ |
-/ |
+/- |
+/- |
-/ |
+/+ |
Winsor *[42] |
||
|
25 |
q32-qter |
+ |
|
+ |
+ |
+ |
+ |
- |
- |
+ |
+ |
- |
+ |
Schmid [33] |
|
|
26 |
q32-qter |
+ |
|
+ |
+ |
- |
+ |
+ |
+ |
- |
+ |
Schinzel [31] |
|||
|
27 |
q32-qter |
+ |
|
- |
+ |
- |
+ |
|
- |
+ |
- |
+ |
Felding [16] |
||
|
28 |
q32-qter |
- |
|
+ |
- |
- |
+ |
|
+ |
+ |
+ |
- |
+ |
Novales [25] |
|
|
29 |
q32-qter |
+ |
+ |
+ |
+ |
+ |
+ |
|
+ |
- |
+ |
- |
+ |
Scelsa [30] |
|
|
30 |
q33-qter |
- |
+ |
+ |
+ |
- |
+ |
|
+ |
- |
+ |
- |
+ |
Bartsch [8] |
|
|
31 |
q33-qter |
- |
- |
+ |
+ |
- |
- |
- |
- |
+ |
- |
+ |
Turleau [36] |
||
|
32 |
q34-qter |
+/+ |
+/- |
-/+ |
+/+ |
-/+ |
+/+ |
-/- |
-/- |
+/+ |
-/+ |
-/- |
+/+ |
Romain*[28] |
|
|
33 |
q36-qter |
- |
+ |
- |
- |
- |
- |
+ |
- |
- |
- |
+ |
Verma [37] |
||
* - клиника двух пациентов с идентичной трисомией 7q; «+» - признак присутствует; «-» - признак отсутствует
Наиболее близкими случаями трисомии 7q, перекрывающими участок 7 q22-q32, как у нашего пробанда, являются 7q22-q31. Мы не обнаружили страбизм, маленький нос, переднюю борозду. Однако скелетные аномалии и макроглоссия были выявлены только у пробанда, описанного в нашей работе. Специфическими особенностями нашего случая можно считать длинное тело при рождении, тугоухость и частичную атрофию зрительных нервов, свидетельствующих об органических поражениях мозга.
Таким образом, можно предположить, что у ребёнка с хромосомной аномалией, впервые описанной в настоящей работе, представлен новый вариант хромосомной патологии, обусловленный дополнительным фрагментом 7q22-q32, транслоцированным в длинное плечо хромосомы 1 и ставшим причиной частичной трисомии хромосомы 7 по этому локусу. Необходимо также отметить, что в работе Cai с соавторами в 2001 году был описан здоровый мужчина - носитель инсерции q22-q32, чей кариотип осложнён дополнительной перестройкой хромосомы 7 в виде транслокации (сбалансированный кариотип) [12]. Его семейный анамнез был отягощён четырьмя спонтанными абортами и двумя живорождениями с летальным исходом через несколько минут после рождения, а хромосомный статус плодов не был установлен. Поэтому невозможно сделать выводы об аберрациях, имевшихся у них и приведших к такому результату. В то же время инсерция материала хромосомы 7 (7q22-q32) в хромосому 1, вызвавшая хромосомный дисбаланс у нашего пробанда, передана ему матерью. Однако при кариотипировании матери у неё была обнаружена еще дополнительная хромосомная аберрация (инверсия), затронувшая уже перестроенную хромосому 7. Поэтому генный состав деривативной хромосомы 7 у матери характеризуется, по-видимому, одновременной потерей и перемещением участка 7q22-q32 в хромосому 1 с инверсией длинного плеча хромосомы 7. Можно предположить, что столь сложная структурная перестройка могла возникнуть поэтапно. Однако сказать, какая аберрация первична, не представляется возможным. Мать, носительница столь сложной хромосомной перестройки, является единственным ребёнком в семье. Учитывая сложность выявленной структурной хромосомной патологии, можно предположить её сегрегацию из поколения в поколение в данной семье. В семейном анамнезе у матери, наряду с рождением дочерей, зарегистрированы два случая самопроизвольного прерывания беременности. Вероятно, это связано с несбалансированным кариотипом при наследовании деривативной хромосомы 7. Столь сложные структурные сбалансированные хромосомные перестройки родителей, представленные выше, редки. По литературным данным, дети, как правило, наследуют частичную трисомию 7q от матери или отца, носителей сбалансированной транслокации, реже инсерции или инверсии [17; 25; 30; 32; 41]. Родители независимо от пола, предположительно с одинаковой вероятностью, могут оказаться носителями подобных аберраций. Появление частичной трисомии 7q также возможно de novo, например при удвоении (дупликации) одного из фрагментов длинного плеча этой хромосомы (табл. 2).
Таблица 2. Варианты хромосомных перестроек с частичной трисомией 7q.
|
Поряд-ковый номер |
Сегмент |
Кариотип пробанда с частичной трисомией 7q |
Методы исследования |
Автор |
|
1 |
7(q31-qter) |
46,XY,der(14)t(7;14)(q31;qter)pat |
G-окрашивание |
Alfi et al. [6] |
|
2 |
7(q31-qter) |
46,XY,der(5)t(5;7)(q35;q31) pat |
G-окрашивание |
Al Saadi et al [7] |
|
3 |
7(q31-qter) |
46,XX,der(9)t(7;9)(q31;p24)mat |
G -окрашивание |
Berger et al. [11] |
|
4 |
7(q21-qter) |
46,XX,der(4)t(4;7)(q35;q21.2)pat |
G-окрашивание FISH |
Courtens et al. [14] |
|
5 |
7(q32-qter) |
46,XX,der(12),t(7;12)(q32 ;qter)pat |
G -окрашивание |
Couzin et al. [15] |
|
6 |
7(q32-qter) |
46,XY,der(18),t(7;18)(q32;q23)mat |
G-окрашивание |
Couzin et al. [15] |
|
7 |
7(q22-q31)
|
46,XX,dup(7)(q22;q31.3) de novo |
G-окрашивание, FISH,CGH |
Megarbane et al. [26] |
|
8 |
7(q32-qter) |
46,XY,der(2),t(2;7)(q37 –q32)pat |
G-окрашивание |
Schmid et al. [33] |
|
9 |
7(q32-qter) |
46,XY,der(5)t(5;7)(q35;q32) de novo |
G-окрашивание, FISH |
Scelsa et al. [30] |
|
10 |
7(q21.2-q31.31) |
46,XX,der(10)t(9;10)(p13;q23),der(9)ins(9;7) (p13;q21.2-q31.31)mat |
G-окрашивание FISH,CGH |
Weimer et al. [41] |
|
11 |
7(q22-q32) |
46,XX, der(1) ins(1;7)(q32;q22q32)mat |
G-окрашивание, FISH |
Настоящий случай |
Обнаруженная нами частичная трисомия 7q22- q32, у ребёнка с умственной отсталостью и аномалиями развития представляется уникальным случаем как с учётом точек разрыва, формирующих сегмент, так и семейной хромосомной перестройки. Выявление сбалансированной аберрации у матери больного ребенка в дальнейшем определит тактику медико-генетического консультирования данной семьи в случае повторной беременности.
Таким образом, использование цитогенетического анализа и FISH-диагностики у пациента с аномалиями развития и умственной отсталостью позволило точно определить сложную хромосомную патологию и обосновать необходимость молекулярно-цитогенетичеcкой диагностики подобных случаев. При клинико-генетическом анализе данной аберрации и сравнении с 35 ранее опубликованными случаями частичной трисомии 7q мы не нашли ясных корреляций фенотип-генотип, которые помогли бы вычленить синдромальные формы частичных трисомий хромосомы 7 (7qàter). Однако необходимо отметить, что объективное сравнение генетических особенностей и уточнение точек разрыва с идентификацией всех генов, вовлеченных в аберрации, возможно лишь при использовании современных технологий полногеномного сканирования на основе ДНК-микроматриц (array CGH) [4; 20]. Поэтому дальнейший молекулярный анализ ранее описанных случаев частичных трисомий 7q позволит более детально определить особенности генетических нарушений в каждом конкретном случае этой редкой хромосомой патологии.
Библиографическая ссылка
Симонова В.В., Ворсанова С.Г., Колотий А.Д., Пинелис В.Г. ЧАСТИЧНАЯ ТРИСОМИЯ 7Q22-Q32: ОПИСАНИЕ РЕДКОГО СЛУЧАЯ ХРОМОСОМНОЙ АНОМАЛИИ И ОБЗОР ЛИТЕРАТУРЫ // Современные проблемы науки и образования. 2013. № 3. ;URL: https://science-education.ru/ru/article/view?id=9229 (дата обращения: 30.06.2026).