



Введение
По данным федерального регистра, численность пациентов с сахарным диабетом (СД) первого типа (СД1), состоящих на диспансерном учете на 21.04.2024 г., составила 292,2 тысячи человек, или 5,6% от всех зарегистрированных в РФ пациентов с СД. СД является одной из основных причин развития хронической болезни почек (ХБП). При СД1 ХБП встречается у 25–75% больных и остается ведущим микрососудистым осложнением, частота нефропатии при СД1 в 2023 г. составила 25,9%, в 2016 г. – 23% [1]. Пожизненный риск заболевания почек при СД1 традиционно оценивается примерно в 50%, но может превышать 70%. Средний возраст дебюта ХБП у лиц с СД1 составляет 40,4 года, длительность заболевания до момента диагностики ХБП – 14,2 года. В структуре смертности пациентов с СД1 терминальная стадия ХБП составляет 5,6%, более 70% пациентов с диабетической болезнью почек (ДБП) умирают в течение пяти лет [2]. Кроме этого, ХБП при СД усложняет лечение и повышает смертность, связанную с сердечно-сосудистыми заболеваниями, новообразованиями, инфекциями, включая вирус иммунодефицита человека и тяжелый острый респираторный синдром при новой коронавирусной инфекции [3].
Накоплено значительное количество экспериментальных и клинических данных, подтверждающих, что окислительный стресс (ОС) прямо или косвенно играет роль в патогенезе, инициировании и прогрессировании хронических осложнений при СД, включая ХБП [4]. Гипергликемия может быть как причиной, так и следствием ОС, который вызывает инсулинорезистентность у пациентов с СД, приводит к дисфункции β-клеток, что усложняет патогенез СД [5]. Использование определенных, обоснованных с патогенетических позиций биомаркеров ОС может иметь значение в прогнозе прогрессирования ХБП, возникновении ее осложнений, оценке эффективности терапии и профилактики. В литературе данная проблема преимущественно освещается при СД второго типа (СД2) с учетом его большей распространенности, однако СД1 встречается у молодых, потенциально трудоспособных пациентов, скорость развития осложнений у них выше, чем при СД2, а их патогенез, включая ХБП, изучен недостаточно, данные о роли про- и антиоксидантных факторов при СД1 противоречивы и разрознены. Понимание молекулярных и клеточных механизмов патогенеза ХБП у пациентов с СД1, связанных с ОС, имеет решающее прикладное значение для разработки маркеров диагностики, прогрессирования, контроля терапии и профилактики, а также эффективных терапевтических подходов.
Цель исследования – провести критический анализ данных,посвященных роли окислительного стресса в патогенезе хронической болезни почек у пациентов с сахарным диабетом первого типа.
Материалы и методы исследования
Для проведения систематического обзора в базах данных Pubmed, РИНЦ, Medline произведен поиск соответствующих исследований за 2017–2024 гг. по ключевым словам: «сахарный диабет первого типа», «хроническая болезнь почек», «диабетическая болезнь почек», «окислительный стресс», «патогенез», «маркеры окислительного стресса». Всего было проанализировано 268 источников литературы, из них в статье использовано 50. Авторы руководствовались современными принципами подготовки обзоров [6].
Результаты исследования и их обсуждение
Окислительный стресс и его роль в организме. ОС возникает, когда баланс между про- и антиоксидантной системами изменяется в пользу первой за счет образования активных форм кислорода (АФК) и азота (АФА) и/или угнетения антиокислительной защиты (АОЗ) [7]. К наиболее значимым АФК относятся супероксид-анион, гидроксильный радикал, диоксид водорода, оксид азота (II). Взаимодействие свободных радикалов друг с другом приводит к образованию нерадикальных соединений, а их атака на нерадикальные молекулы запускает цепные реакции. Примерами нерадикальных окислителей являются перекись водорода, озон, синглетный кислород, хлорноватистая кислота, пероксинитрит, перекиси липидов и др. Избыточная продукция АФК возникает в результате активации ферментных систем, таких как семейство NOX (NADPH-оксидаза), комплексы I и III электронтранспортной цепи митохондрий, монооксигеназы (при участии цитохром P450), ксантиноксидаза, синтаза оксида азота (II), миелопероксидаза. ОС возникает на фоне нарушения АОЗ, включая ферментативные (активность супероксиддисмутазы (СОД), каталазы, глутатионпероксидазы (ГПО), глутатионредуктазы) и неферментативные (липоевая кислота, глутатион, L-аргинин, коэнзим Q10, мелатонин, мочевая кислота, билирубин, трансферрин и др.) эндогенные пути. Экзогенные, поступающие в организм с продуктами питания, антиоксиданты включают витамины Е и С, каротиноиды, ликопин, микроэлементы селен, марганец, цинк, флавоноиды, омега-3 и омега-6 жирные кислоты.
Двойственную роль в организме играют АФК и АФА: в небольших концентрациях (окислительный эустресс) они выступают как вторичные мессенджеры, регулируя рост, дифференцировку клеток, апоптоз, удаление опухолевых клеток, патогенных агентов при воспалении, детоксикацию ксенобиотиков, синтез простагландинов и лейкотриенов и иное (рис. 1); при высоких концентрациях (окислительный дистресс) прооксиданты изменяют внутриклеточную сигнализацию через пути Nrf2/Keap1 и NF-κB, инактивируют ферменты гликолиза, цикла Кребса и электронтранспортной цепи митохондрий, становятся токсичными, вызывая повреждение биомолекул (липидов, белков и нуклеиновых кислот), и в конечном итоге клеток и тканей, что нарушает функционирование организма в целом [8]. Пероксидация липидов изменяет проницаемость и свойства мембран клетки, окисление ядерной или митохондриальной ДНК приводит к разрыву нитей, аберрантному сшиванию и образованию аддуктов ДНК, окисление белков (включая важнейшие ферменты) вызывает потерю их биологических свойств. Доказана роль ОС при старении, в патогенезе СД, неоплазии, заболеваний воспалительного и аутоиммунного генеза, атеросклероза, ишемической болезни сердца, цереброваскулярной патологии и др. [9].
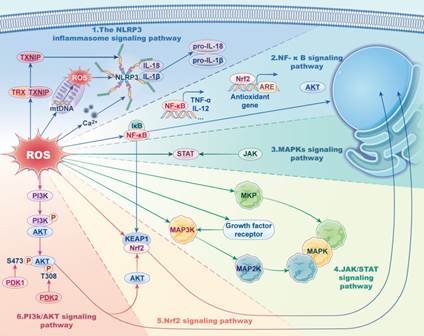
Рис. 1. АФК как вторичные мессенджеры в регуляции воспаления через сигнальные пути: NLRP3, NF-κB, MAPKs, JAK/STAT, сигнальный путь Nrf2 и PI3K/AKT [10]
Патогенез окислительного стресса при СД является сложным и многофакторным, ключевой механизм – изменение концентрации глюкозы в плазме, при этом имеют значение как стойкая гипергликемия (верифицируемая по определению HbA1c), так и колебания (вариабельность) гликемического профиля и гипогликемия в условиях терапии СД [8]. При гипергликемии избыточная генерация АФК обусловлена активацией окисления глюкозы в цикле Кребса и, как следствие, потока электронов в составе НАДН и ФАДН2 в электронтранспортную цепь митохондрий, где после достижения критического порога электрохимического потенциала Н+ избыток электронов взаимодействует с О2 с образованием супероксид-анион радикала [11]. При гипергликемии снижается активность ключевого гликолитического фермента глицеральдегид-3-фосфатдегидрогеназы с последующим накоплением глицеральдегид-3-фосфата и его метаболитов – метилглиоксаля и диацилглицерола. В свою очередь, метилглиоксаль является источником конечных продуктов гликирования (AGE), а диацилглицерол активирует протеинкиназу С. AGE образуются в результате неферментативных реакций (реакция Майяра) между карбонильными группами глюкозы, галактозы, рибозы и фруктозы, продуктами метаболизма глюкозы (рибозо-5-фосфат, глюкозо-6-фосфат, глицеральдегид, фруктозо-6-фосфат, дезоксирибоза-5-фосфат) с аминогруппой и образованием продуктов Амадори, а затем метилглиоксаля, глиоксаля и малонового диальдегида [12]. AGE после взаимодействия с рецепторами на мембране (RAGE), а также протеинкиназа С через активацию NF-κB увеличивают синтез цитокинов, хемокинов, адгезивных молекул для лейкоцитов, НАДФН-оксидазы, что приводит к эскалации ОС. AGE запускают каскад внутриклеточных путей МАРК, JNK, фосфоинозитид-3-киназы, ингибируют антиоксидантные системы и нарушают гомеостаз железа, инициируя ферроптоз клеток [13]. Протеинкиназа С активирует другие ферменты прооксидантного статуса – синтазу оксида азота (NOS), ксантиноксидазу (XO) и липоксигеназу В [14]. В условиях гипергликемии активируется полиоловый путь метаболизма глюкозы с образованием сорбитола и снижением внутриклеточного содержания NADPH – кофактора глутатионредуктазы, необходимой для восстановления глутатиона и реализации антиоксидантного потенциала глутатионпероксидазы. Дальнейший метаболизм сорбитола до фруктозы сопряжен с образованием фруктозо-6-фосфата, фруктозо-3-фосфата и далее 3-дезоксиглюкозы и 3-дезоксиглюкозона, взаимодействие которых с аминогруппами белков приводит к формированию AGE. Фруктозо-3-фосфат включается в гексозаминовый путь метаболизма с образованием глюкозамин-6-фосфата, гликозилированием белков, нарушением гомеостаза кальция и генерацией АФК как отражение стресса эндоплазматического ретикулума. Роль гипергликемии и сопряженных метаболических путей в патогенезе ОС при СД резюмирована на рисунке 2 [14].
Вариабельность гликемии с эпизодами гипер- и гипогликемии различной частоты и выраженности, нередко наблюдаемая у больных с СД1, связана с патогенезом ОС за счет активации НАДФН-оксидазы с образованием супероксид-анион радикала и повреждением микрососудов, формированием нефропатии [15]. Вариабельный гликемический профиль при СД приводит к поляризации макрофагов в М1 провоспалительный фенотип через сигнальный путь TLR4-IRF5, секреции IL-1β, TNF-α, IL-6, моноцитарного хемоаттрактантного белка-1 и, как следствие, увеличению генерации АФК фагоцитами [16]. Средняя амплитуда и стандартное отклонение уровня гликемии при СД1 имеют сильную положительную корреляцию с концентрацией в плазме оксистерола – продукта неэнзиматического окисления холестерина и надежного биомаркера ОС [17]. Эпизоды гипогликемии у больных СД1 представляют не меньшую опасность в отношении формирования ОС за счет активации симпатоадреналовой системы и выделения катехоламинов, которые вызывают ишемию и гипоксию тканей с последующей реперфузией и, как следствие, активацию свободнорадикального окисления, повышают активность тромбоцитов, их секрецию АФК и вклад в ишемию тканей [18]. В условиях гипогликемии формируется лейкоцитоз, увеличиваются активность фагоцитов и НАФН-зависимая генерация АФК, синтез и секреция провоспалительных цитокинов. При гипогликемии в митохондриях обнаружено увеличение АФК, а в крови накапливаются продукты ПОЛ.
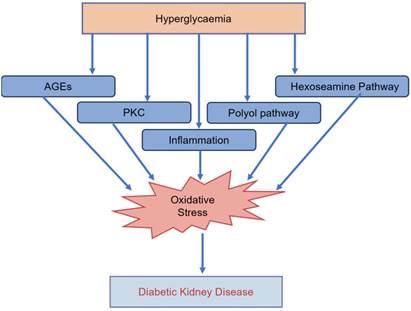
Рис. 2. Роль гипергликемии и сопряженных метаболических путей в патогенезе ОС при СД [14]
Провоспалительный статус при СД реализуется за счет рецептор-опосредованной активации AGE внутриклеточных сигнальных путей NF-κB, Nrf2, MAPK и иных, что приводит к аккумуляции, активации лейкоцитов, секреции ими провоспалительных цитокинов (IL-1β, TNF-α, IL-6, IL-23, MCP-1, MIP-1β, ГМ-КСФ и др.), АФК, АФА и отражает феномен оксивоспаления (oxi-Inflammation) при СД, когда ОС и воспаление потенцируют друг друга в условиях гипергликемии и инсулинорезистентности и участвуют в патогенезе осложнений при СД (рис. 3) [19]. Показана роль NOD-подобного рецептора (NLR) в реализации воспаления при СД за счет синтеза цитокинов и повреждении почек [20]. Таким образом, при СД генерация АФК ассоциирована с провоспалительными изменениями гомеостаза, активностью НАДФН-оксидазы (особенно изоформы NOX4) и протеинкиназы С, полиоловым и гексозаминовым путями метаболизма глюкозы, образованием AGE.
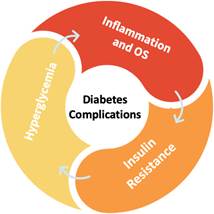
Рис. 3. Порочные круги ОС, воспаления, инсулинорезистентности и гипергликемии в патогенезе осложнений при СД [19]
В патогенезе ОС при СД, помимо усиления генерации АФА и АФА, имеет значение снижение активности АОЗ. Так, активация полиолового пути метаболизма глюкозы при СД ассоциирована не только с увеличением продукции АФК, но и со снижением активности ферментов АОЗ, таких как супероксиддисмутаза и каталаза. При СД гипергликемия приводит к гликированию и последующей инактивации СОД, а полиоловый путь метаболизма глюкозы – к истощению NADPH и снижению активности ГПО и глутатионредуктазы [11]. До определенного момента формирование ОС при СД сдерживается активацией компонентов АОЗ редокс-чувствительным транскрипционным ядерным эритроид-2-подобным фактором (Nrf2) и FoxO, что ограничивает повреждение подоцитов, клеток мезангия, фиброгенез в почках при СД [21]. Nrf2 активирует экспрессию генов факторов АОЗ: Cu, Zn-СОД, пероксиредоксинов, тиоредоксинов, ГПО/глутатионредуктазы, глутатион-S-трансферазы, глутаматцистеинлигазы, гемоксигеназы-1 и др. Определенное значение в активации АОЗ при СД имеет система факторов транскрипции FoxO [22]. Установлена роль однонуклеотидных полиморфизмов генов антиоксидантов Mn-СОД, каталазы и ГПО в патогенезе ОС при СД и диабетической нефропатии [23]. В условиях СД эскалация ОС связана со снижением активности ферментов АОЗ за счет эпигенетических изменений – метилирования ее ДНК [24]. Однако роль эпигенетических изменений компонентов редокс-статуса при СД, включая метилирование ДНК, модификацию гистонов и хроматина, некодирующие РНК, дискутабельна: они могут быть как причиной, так и следствием изменений гомеостаза [25]. Примечательно, что некоторые эпигенетические изменения, вызванные гипергликемией, сохраняются в клетках-мишенях даже после нормализации глюкозы, что поддерживает ОС при СД, несмотря на гликемический контроль, и рассматривается как метаболическая память при диабетической нефропатии [26].
Роль окислительного стресса в патогенезе ХБП при СД. Накопление AGE, продуктов полиолового и гексозаминового путей метаболизма глюкозы, активация НАДФН-оксидазы и протеинкиназы С, снижение активности АОЗ, ассоциированные с ОС при СД, приводят к формированию низкоинтенсивного системного воспаления, гемодинамическим, метаболическим изменениям, активации синтеза соединительной ткани, которые, в свою очередь, запускают формирование ДБП и соответствующие патоморфологические изменения: воспаление в почечной ткани, расширение и изменение тонуса сосудов мезангия, гипертрофию на уровне клубочкового аппарата, нефрофиброз и гломерулосклероз [21]. При ОС в условиях СД первыми мишенями для АФК и АФА в почках выступают базальные мембраны и клетки мезангия, эндотелиоциты, подоциты [27].
Центральным звеном инициации ДБП выступает опосредованное ОС повреждение подоцитов, запускающее апоптоз, некроз, аутофагию, пироптоз, ферроптоз, купроптоз и подоптоз (рис. 4) [28].
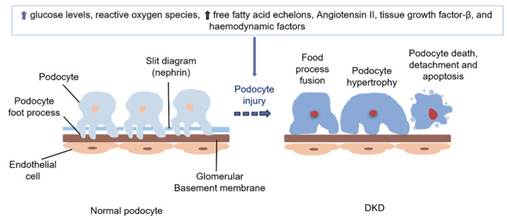
Рис. 4. Патофизиологические аспекты повреждения подоцитов при СД [21]
Одним из механизмов необратимого повреждения подоцитов в патогенезе диабетической нефропатии выступает стресс эндоплазматического ретикулума (ЭПР) – связующее звено ОС, воспаления и митохондриальной дисфункции [29].
Индукторами стресса ЭПР выступают гипергликемия, свободные жирные кислоты, окисленные липопротеины и другие продукты ОС, а его последствия в виде нарушения конформации белков и выхода Са2+ вызывают митохондриальную дисфункцию и повышение генерации АФК, замыкая таким образом порочный круг.
Почки являются постоянно функционирующим активным органом, клетки которого богаты митохондриями, что делает их уязвимыми для повреждений, вызванных ОС при СД, ускорения формирования ХБП [11]. Плотность митохондрий в почке является одной из самых высоких в организме, что связано с высокими энергетическими потребностями для реализации функций [30]. У пациентов с СД наблюдается дисфункция митохондрий в почках, что рассматривается не только как ключевой механизм ОС, но и как фактор патогенеза диабетической нефропатии [31]. Данные многих исследований свидетельствуют о сильной связи между возникновением и прогрессированием ДБП и глюкозо-индуцированной и не связанной с гипергликемией митохондриальной дисфункцией [32]. Кроме гипергликемии, факторами митохондриальной дисфункции выступают свободные жирные кислоты, уровень которых при СД повышается. В условиях гипергликемии при СД в клетках почек зафиксирован эффект Варбурга с подавлением митохондриального дыхания и переключением метаболизма глюкозы на анаэробный путь, что, в свою очередь, ассоциировано с активацией метаболических путей с образованием сорбитола и гексозамина – источников AGE [14, 33]. Как следствие митохондриальной дисфункции снижается синтез АТФ и увеличивается генерация АФК – молекулярные механизмы повреждения подоцитов и прогрессирования диабетической нефропатии [24]. Кроме подоцитов, показана роль митохондриальной дисфункции при СД в повреждении эпителиоцитов канальцев нефрона, эндотелиоцитов почечных капилляров, иммунных клеток в почках [34].
Локальное воспаление в почках, инициируемое АФК, АФА, AGE и иными факторами, запускает повреждение и гибель клеток, связано с активацией ренин-ангиотензин-альдостероновой системы, что потенцирует развитие ДБП и ухудшение функции почек. Триада ОС, хронического воспаления (оксивоспаления) и эндотелиальной дисфункции при СД формирует еще один порочный круг хронического повреждения в почках, усугубляет существующие нарушения и приводит к системным осложнениям, включая дисфункцию сердечно-сосудистой системы [35]. ОС при СД вызывает эндотелиальную дисфункцию, повышение проницаемости сосудов, активацию тромбообразования, дестабилизацию микроциркуляции в почках, что усугубляет ишемические повреждения, приводит к прогрессированию ХБП [36]. ОС влияет на метаболизм липидов: у пациентов с СД1 изменяется липидный профиль, повышается образование окисленных липопротеинов низкой плотности (oxLDL), которые потенцируют воспаление в почках [37]. Гипергликемия, AGE, ангиотензин II и альдостерон повреждают паренхиматозные клетки почек, вызывают их дисфункцию, поддерживают хроническое воспаление [38].
В условиях хронического оксивоспаления в почках формируются гломерулосклероз, атрофия клеток канальцев, интерстициальный фиброз, альбуминурия и прогрессирующая утрата функции почек, а ОС рассматривается как мишень ограничения нефрофиброза при ДБП [21, 39]. С активацией при СД протеинкиназы С связаны синтез фактора роста фибробластов, фактора роста эндотелия сосудов, TGF-β1, фибронектина, коллагена IV типа, которые играют решающую роль в развитии нефрофиброза [40].
Ряд исследователей акцентируют внимание на TGF-β1 в патогенезе диабетической нефропатии [41]. Патоморфологическим эквивалентом поражения почек при СД выступает индуцируемый фиброгенными медиаторами, такими как TGF-β1, сигнальными путями TGF-β/Smad, Wnt/β-catenin и Notch, а также АФК, продуктами ОС AGE эпителиально-мезенхимальный переход со снижением экспрессии эпителиальных клеточных маркеров, например Е-кадгерина, и повышением фибронектина, виментина, фибробласт-специфического белка-1 (α-SMA, S100A4) и других мезенхимальных маркеров [42].
Маркеры окислительного стресса при ХБП у больных СД1. Расчетная скорость клубочковой фильтрации (рСКФ) и альбуминурия являются общепризнанными и наиболее часто используемыми маркерами утраты почечной функции, однако в настоящее время накапливаются данные, ставящие под сомнение их надежность и ограниченность применения при ДБП [43]. Так, при начальных стадиях ХБП при СД1, а по некоторым данным – у 30% пациентов с СД, не наблюдается альбуминурия вплоть до терминальной ХБП, в связи с чем сформировалось представление о неальбуминурической форме ДБП [44].
У части больных с ДБП может быть регрессия альбуминурии, хотя обычно альбуминурия ассоциирована со снижением рСКФ [45]. При этом принципиально важно для начала терапии раннее, до появления альбуминурии, выявление поражения почек при СД. Более информативным для диагностики ДБП является индекс в моче альбумин/креатинин [46]. Как и альбуминурия, рСКФ также имеет важные ограничения в качестве прогностического биомаркера ХБП при СД [47]. Не достигнуто единого мнения о скорости снижения рСКФ при СД [46]. Учитывая феномен гиперфильтрации на начальном этапе ДБП, снижение рСКФ не отражает изменение количества функционирующих нефронов. При этом нормоальбуминурическая гиперфильтрация является фактором риска быстрого снижения функции почек при СД1.
Таким образом, существует острая необходимость в новых биомаркерах дисфункции почек при СД для надежного прогнозирования ее возникновения и прогрессирования [48]. В этом отношении перспективны метаболиты редокс-статуса, учитывая многофакторный патогенез ОС при ХБП в условиях СД1, перекрестные взаимодействия ОС, воспаления, эндотелиальной дисфункции, нарушения микроциркуляции и гемодинамики. Большинство исследователей полагают, что совокупность биомаркеров представляет большую информативность, диагностическую и прогностическую ценность при диабетической нефропатии, чем единичные биомаркеры [41].
Из-за нестабильности и короткого времени жизни АФК и АФА их прямое измерение затруднительно, поэтому для оценки редокс-статуса используют долгоживущие продукты окисления. В качестве маркеров ОС могут выступать AGE, 3-дезоксиглюкозон, метилглиоксаль, метионинсульфоксид, 2-аминоадипиновая кислота, которые продемонстрировали прогностическую значимость в отношении прогрессирования ХБП при СД. Биомаркерами ОС при ХБП у больных с СД1 выступают миелопероксидаза, которая была связана с высоким уровнем HbA1c (9,6%), а также Nrf2 [48].
Повышение уровня митохондриальной ДНК в крови и в моче отражает дисфункцию митохондрий и тяжесть поражения почек при СД [49]. Однако из-за сложных методов, необходимых для детекции указанных факторов, они редко используются на практике, в этом отношении представляют интерес такие маркеры ОС в сыворотке, моче и иных биологических средах, как продукты перекисного окисления липидов (4-гидроксиноненаль, малоновый диальдегид, 8-изопростан, диеновые конъюгаты, кетодиены и сопряженные триены), окисления ДНК (8-OH-дезоксигуанозин (8-OHdG)) и продукты окислительной деструкции (карбонилирования) и окисления белков (расширенный спектр альдегид- и кетондинитрофенилгидразонов) [50].
Информативным маркером ОС при диабетической нефропатии является концентрация в моче 8-OHdG, его высокий уровень ассоциирован с прогрессированием ХБП и смертностью при СД1 [42]. Показаны предикторная роль расширенного спектра маркеров окислительной деструкции (карбонилирования) белков в отношении дебюта ХБП при СД, положительная корреляция их уровня в крови с концентрацией глюкозы, HbA1с, креатинина, СКФ. Среди антиоксидантнов установлена прогностическая роль поражения почек при СД1 в отношении СОД, а также общей антиоксидантной активности плазмы. При ДБП биомаркеры воспаления позволяют опосредованно судить об интенсивности ОС и проводить скрининг поражения почек, к их числу относят TNF-α и его рецепторы TNFR-1, TNFR-2, IL-6, IL-18, нейтрофильный желатиназно-ассоциированный липокалин (NGAL), катепсин S макрофагального происхождения, С-РБ, церулоплазмин, Е-селектин, моноцитарный хемоаттрактантный белок-1, sP-селектин, нейтрофильные внеклеточные ловушки [39, 47].
Заключение.ОС является результатом дисбаланса между про- и антиоксидантной системами, проявляется избыточным образованием АФК, АФА, угнетением АОЗ и лежит в основе патогенеза многих заболеваний, в том числе СД. При СД в условиях гипергликемии или вариабельности гликемического профиля патогенез ОС включает активацию полиолового и гексозаминового путей метаболизма глюкозы, образование AGE, активацию протеинкиназы С, формирование провоспалительного статуса с активацией НАДФН-оксидазы, миелопероксидазы, ксантиноксидазы, NO-синтазы и иных прооксидантных ферментов, снижение синтеза и активности, в том числе за счет полиморфизма генов и эпигенетических изменений ферментов АОЗ – Cu, Zn- и Mn-супероксиддисмутазы, каталазы, глутатионпероксидазы, глутатионредуктазы, гемоксигеназы-1 и др. В условиях ОС при СД в почечной ткани происходит повреждение клеток и компонентов интерстиция, запускающее апоптоз, некроз, аутофагию, пироптоз, ферроптоз, купроптоз, стресс эндоплазматического ретикулума, дисфункцию митохондрий, формируются эндотелиальная дисфункция, оксивоспаление; наблюдаются расширение и изменение тонуса сосудов мезангия, гипертрофия на уровне клубочкового аппарата, эпителиально-мезенхимальный переход, разрастание соединительной ткани за счет эффектов фактора роста фибробластов, фактора роста эндотелия сосудов, TGF-β1, ангиотензина-II и иных факторов, что проявляется прогрессирующим снижением СКФ и альбуминурией. Маркеры ОС могут быть полезны для раннего выявления, мониторинга терапии и профилактики, прогноза прогрессирования и исходов ХБП при СД1. К числу наиболее информативных и доступных в клинической практике относят определяемые в крови и моче продукты окислительной деструкции липидов (малоновый диальдегид, 8-изопростан, диеновые конъюгаты, кетодиены и сопряженные триены), ДНК (8-OH-дезоксигуанозин), белков (расширенный спектр альдегид- и кетондинитрофенилгидразонов), а также показатели выраженности воспалительного процесса (TNF-α и его рецепторы, IL-6, С-РБ, церулоплазмин и др.).
Библиографическая ссылка
Осиков М.В., Журавлева Л.Ю., Эфрос Л.А., Федосов А.А. РОЛЬ ОКИСЛИТЕЛЬНОГО СТРЕССА В ПАТОГЕНЕЗЕ ХРОНИЧЕСКОЙ БОЛЕЗНИ ПОЧЕК У ПАЦИЕНТОВ С САХАРНЫМ ДИАБЕТОМ ПЕРВОГО ТИПА // Современные проблемы науки и образования. 2024. № 6. ;URL: https://science-education.ru/ru/article/view?id=33837 (дата обращения: 29.06.2026).
DOI: https://doi.org/10.17513/spno.33837