



Введение
Современные методики борьбы с воспалительными процессами в рамках лечения мышечной дистрофии Дюшенна, использующие кортикостероиды, несмотря на способность тормозить прогрессирование патологии, ограничены в своей эффективности и часто провоцируют нежелательные реакции. Альтернативой традиционным методам, предоставляющим кратковременное улучшение, выступает этиотропная стратегия, которая направлена на устранение корневых причин недуга, в особенности коррекцию мутации гена DMD. Достижение коррекции уровня дистрофина рассматривается как один из перспективных вариантов терапии [1].
Цель исследования – изучить современные терапевтические методы генной терапии миодистрофии Дюшенна, перспективные разработки в области вирусной генной терапии, рассмотреть эффективность нового препарата SPR-9001 в лечении миодистрофии Дюшенна.
Материал и методы исследования
Для написания обзора использовались электронные поисковые системы: PubMed, Embase, Cochrane Library до ноября 2023 г. Поиск проводился с использованием ключевых слов: «мышечная дистрофия Дюшенна», «дистрофин», «аденоассоциированные вирусы», SPR-9001. Обзор составлен в соответствии с рекомендациями PRISMA (Preferred Reporting Items for Systematic reviews and Meta-Analyses) [2].
Результаты исследования и их обсуждение
Мышечная дистрофия Дюшенна – генетически детерминированное заболевание, сцепленное с Х-хромосомой, клинически характеризующееся прогрессирующей мышечной слабостью, с частотой 1 на 3500–6000 рожденных мальчиков. Это вызвано мутацией гена DMD, который кодирует дистрофин, субсарколеммальный белок, необходимый для структурной стабильности мышц. Генетические дефекты гена делятся на: делеции (65%), дупликации (5–10%) и точечные мутации (10–15%). В настоящее время не существует радикального лечения. Симптомы проявляются в детском возрасте (ограничение подвижности и ранние респираторные осложнения). К основным признакам и симптомам относятся нарушения походки, начинающиеся в раннем детстве, с более поздним появлением нарушений функций дыхания и сердца, что напрямую влияет на прогноз [3]. Респираторные осложнения сокращают продолжительность жизни пострадавших. Не существует лечения, которое изменило бы эволюцию заболевания, хотя кортикостероиды и новые методы генной терапии уменьшают прогрессирование заболевания [4]. У людей с мышечной дистрофией Дюшенна развивается слабость дыхательных мышц, что приводит к нарушению клиренса дыхательных путей и со временем – к дыхательной недостаточности и смерти. Оценка эффективности кашля по жизненной емкости легких, пиковой скорости кашля и максимальному давлению на вдохе и выдохе использовалась для определения оптимального времени начала применения препаратов для терапии кашля [5]. Большинство пациентов становятся тяжелыми инвалидами, нуждающимися в постороннем уходе. Типичными проблемами, возникающими при длительной неподвижности, являются пролежни, тромбозы венозной системы нижних конечностей и аспирационная пневмония. В качестве предикторов возникновения тромбоэмболических осложнений в легочной артерии у пациентов с диагностированным тромбозом венозной системы выступают D-димеры и антитромбин III, как было установлено с помощью многофакторного статистического анализа [6, 7]. Нормальное функционирование дыхательной системы, сравнимое с функционированием у их здоровых ровесников, отмечается у младенцев, страдающих мышечной дистрофией Дюшенна. Эти рано выявленные случаи не предполагают необходимости в расширенных диагностических или лечебных процедурах. С появлением тотального скрининга новорожденных и развитием генетической диагностики множество случаев мышечной дистрофии Дюшенна теперь выявляется на самых ранних этапах жизни. В отличие от прошлых десятилетий, когда летальный исход наступал в возрасте 20–30 лет, современные методы искусственной вентиляции значительно продлили ожидаемую продолжительность жизни мальчиков до более чем 40 лет. В возрасте между 18 и 20 годами больные мышечной дистрофией Дюшенна часто начинают использовать неинвазивные способы вентиляции с положительным давлением при появлении признаков дыхательной недостаточности. В возрасте 12–20 лет снижение подвижности ограничивает функцию дыхательных путей, что снова увеличивает риск вторичной пневмонии и дыхательной недостаточности при вирусных заболеваниях. Отсутствие ходьбы провоцирует развитие сколиоза. Обычно это начинается с искривления грудопоясничного отдела и не оказывает существенного влияния на респираторный статус до дальнейшего прогрессирования [8]. В позднем периоде у пациентов наблюдается значительное снижение подвижности как легких, так и грудной клетки. Частично это снижение может быть связано с ателектазом, возникающим при уменьшении объема легких [9-11].
Миодистрофия Дюшенна является генетическим заболеванием, обусловленным мутацией гена дистрофина, расположенного на хромосоме Xp21 [12, 13]. Данный ген наследуется как сцепленный с Х-хромосомой рецессивный признак, мутации приводят к ограниченной продукции белка дистрофина (рисунок), что способствует потере целостности мембраны миофибрилл с повторяющимися циклами некроза и регенерации [14-16].
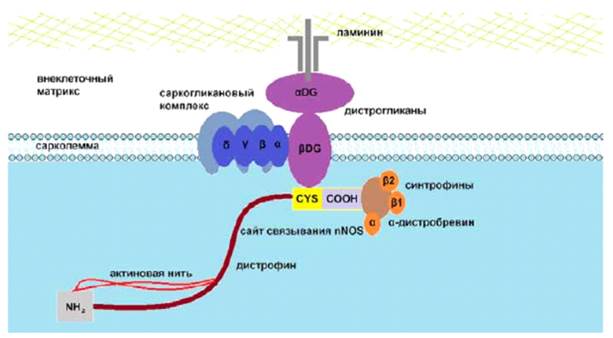
Дистрофин-ассоциированный белковый комплекс [1]
Волокнистая соединительная ткань и жир постепенно замещают мышцы, что приводит к клиническим проявлениям [17, 18]. Ген дистрофина является одним из крупнейших генов в геноме человека. Этот ген, содержащий 79 экзонов и 2,5 Мб ДНК, кодирует белок с молекулярной массой 427 кДа [19]. В настоящее время не существует лекарства от мышечной дистрофии Дюшенна. Тем не менее, были достигнуты успехи в терапии, начиная с внедрения системных кортикостероидов и создания комплексной междисциплинарной помощи, включая физиотерапию и лечение желудочно-кишечных, респираторных и сердечных осложнений. Непрерывное применение кортикостероидов может первоначально задержать прогрессирование заболевания и сохранить некоторые функции, но часто связано с серьезными долгосрочными побочными эффектами. Заместительная терапия дистрофином представляет собой еще одну терапевтическую стратегию при мышечной дистрофии Дюшенна. Недавно управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (Food and Drug Administration (FDA)) одобрило фосфордиамидатные морфолиноолигомеры, способные «пропускать» определенные экзоны гена, чтобы обеспечить выработку укороченного, но функционального дистрофина, для терапии группы пациентов. Этеплирсен был одобрен FDA по ускоренной схеме одобрения для пациентов с мутациями экзона 51 на основании значительного увеличения дистрофина по сравнению с исходным уровнем в течение 48 недель, что с достаточной вероятностью могло предсказать клинический эффект [20]. Последующие исследования этеплирсена подтвердили локализацию дистрофина в сарколемме и его накопление с течением времени и показали функциональное ослабление легочной недостаточности и более длительную ходьбу по сравнению с контрольной группой с совпадением по мутации, а также увеличение выживаемости при непрямом сравнении лечения с использованием стандартной терапии. Из-за специфичности мутаций они не являются вариантом лечения примерно 70% населения с мышечной дистрофией Дюшенна. Генная терапия представляет собой еще одну терапевтическую стратегию лечения, основанную на использовании вирусного вектора для доставки функционального гена в целях компенсации мутировавшего гена с потенциалом длительного эффекта лечения после однократного введения [21]. Генная терапия потенциально может улучшить исход тяжелых заболеваний после однократного введения. Развитие генной терапии требует обширной разработки на каждом этапе: необходимы доклиническая работа по созданию и оценке средств доставки терапии, разработка программ клинических разработок и создание крупномасштабного производственного процесса. Новаторские генные методы лечения вызывают побочные эффекты, поскольку исследователи сталкиваются с множеством проблем, характерных для этого метода лечения.
Следовательно, преимущества этих методов лечения выходят за рамки предоставления знаний о лечении какого-либо одного заболевания, а также создают новые платформы и парадигмы, которые ускорят развитие будущих генных терапий [2, 22, 23]. Открытия в области генной терапии, в том числе стратегии внедрения лекарственных средств с использованием вирусного и невирусного подхода, оказались весьма перспективными для терапии нервно-мышечных патологий и, в частности, для борьбы с мышечной дистрофией Дюшенна [24–26]. Векторы на основе аденоассоциированных вирусов особенно интересуют ученых в контексте разработки новых методов лечения [27–29]. Тем не менее, даже после тщательного системного применения некоторые трудности все еще сохраняются. К ним относятся трудности в проникновении через гематоэнцефалический барьер, агрессивная реакция иммунной системы пациентов, а также риск, что может потребоваться увеличение дозировки. Исследования, направленные на решение этих вопросов, предлагают новаторский метод интратекальной доставки прямо в центральную нервную систему и через спинномозговую жидкость. Ожидается, что применение этого подхода значительно оптимизирует перенос препарата в мозговую ткань, показывая преимущества перед традиционной системной доставкой [30, 31]. Терапевтические стратегии, которые в настоящее время находятся на стадии оценки, включают терапию стволовыми клетками, заместительную ферментную терапию, терапию на основе РНК или ДНК (антисмысловые олигонуклеотиды) и генную заместительную терапию на основе вирусных векторов, включая аденоассоциированные вирусы [32, 33, 34].
Аденоассоциированные вирусы – инструмент в терапии замены гена. С небольшими модификациями нативного вируса они непатогенны и в целом безопасны для человека. Рекомбинантные векторы благодаря своим уникальным серотипам, как естественно встречающимся, так и тем, что были разработаны недавно, обладают потенциалом направленного воздействия на разнообразные ткани. Специфическое сродство этих серотипов к целевым клеточным рецепторам определяет их уникальность. Процесс выборки серотипа очень важен, особенно в контексте их способности трансграничного проникновения гематоэнцефалического барьера. Дополнительным условием для успешной доставки трансгена является точный отбор адаптированного для данного контекста промотора клеток, поскольку он и впоследствии регуляторные элементы играют значительную роль в эффективной экспрессии трансгена внутри клеток-хозяев. Колоссальное влияние на постинтернализационную активность вектора оказывает также композиция его геномной структуры [2, 32].
SRP-9001 является кандидатом на генную терапию для улучшения двигательной функции у людей с мышечной дистрофией Дюшенна. SRP-9001 развертывает аденоассоциированный вирус серотипа rh74 (AAVrh74) для переноса функциональной копии отсутствующего или дефектного гена дистрофина в мышечные клетки. Данный препарат предназначен для доставки гена, кодирующего так называемый белок микродистрофина, который работает аналогично нормальному дистрофину, хотя и имеет меньший размер. В терапии используется безвредный модифицированный вирус (AAVrh74), обладающий высокой аффинностью к мышечной ткани, что позволяет осуществлять адресную доставку. Кроме того, у него есть специфический для мышц промотор – элемент ДНК, контролирующий активность гена, называемого MHCK7, предназначенный для усиления активности гена в сердечных и скелетных мышцах. В клиническом исследовании SRP-9001, проведенном в 2020 году, приняли участие четыре пациента – мальчики 4–7 лет. Препарат вводился пациентам однократно, внутривенно. Все нежелательные явления (n = 53) были расценены как легкие (33 [62%]) или умеренные (20 [38%]), серьезных нежелательных явлений не наблюдалось. Восемнадцать нежелательных явлений считались связанными с лечением, наиболее частым из которых была рвота (9 из 18 явлений [50%]). У трех пациентов было временное повышение уровня γ-глутамилтрансферазы, которое разрешилось кортикостероидами. Через 12 недель иммуногистохимия образцов биопсии икроножной мышцы выявила сильную экспрессию трансгена у всех пациентов, в среднем 81,2% мышечных волокон экспрессировали микродистрофин со средней интенсивностью 96% в сарколемме. Вестерн-блоттинг показал среднюю экспрессию 74,3% без корректировки жира или фиброза и 95,8% с корректировкой. У всех пациентов продемонстрированы функциональное улучшение показателей NSAA и снижение уровня креатинкиназы (после лечения по сравнению с исходным уровнем), которые сохранялись в течение 1 года [35].
Функциональные возможности двигательной системы у мальчиков, страдающих мышечной дистрофией Дюшенна и находящихся под наблюдением в амбулаторных условиях, подверглись оценке с помощью различных тестирований. Измерения включали наблюдение за продолжительностью восхождения по четырем лестницам, фиксацию времени, необходимого для 100-метровой ходьбы, а также применяли интегрированную амбулаторную методику оценки North Star (NSAA) – оценка моторной функции у юных пациентов с мышечной дистрофией Дюшенна. Отмечено значительное улучшение результатов испытаний по истечении 90 дней терапии в сравнении с данными, зарегистрированными до начала терапии. Увеличение на 8,6 балла по шкале NSAA зарегистрировано по прошествии трехлетнего периода после начала применения SRP-9001 в сравнении с изначальными показателями.
Критерии исключения участников из дальнейшего участия в исследовании заключались в следующем:
- наличие проблем в работе почек и печени, определенных согласно методическим указаниям протокола эксперимента;
- использование любого лекарства для пропуска экзона, если до скрининга прошло менее шести месяцев;
- выявление патологий кардиоваскулярной системы при проведении эхокардиографического обследования [23].
Исследование, проведенное компанией для Roche Group, также показало положительный результат в лечении мышечной дистрофии Дюшенна. Промежуточные результаты первых 11 участников когорты 1 показали устойчивую экспрессию микродистрофина и безопасность препарата. За 48 недель применения SRP-9001 участники эксперимента в целом показали повышение NSAA балла (North Star Ambulatory Assessment – шкала оценки функциональных возможностей пациента, состоящая из 17 пунктов, которая служит для оценки функциональных возможностей пациента), однако это увеличение не достигло статистической значимости по сравнению с группой, которой давали плацебо. Результаты данного исследования станут основой в разработке дизайна клинического исследования 3-й фазы. Также было проведено исследование для оценки экспрессии SRP-9001 в течение 260 недель путем внутривенных инфузий, доказывающее безопасность препарата. Участниками стали дети от 3 до 7 лет с мышечной дистрофией Дюшенна, ранее не лечившиеся кортикостероидами или принимающие стабильную дозу, эквивалентную пероральным кортикостероидам, не менее 12 недель. На 30-й день у участников в возрасте до 3 лет была проведена оценка кишечной моторики по специальной шкале Bayley-III, а у участников до 5 лет – 100-метровый тест на время. На 90-й день было проведено количественное сравнение текущего уровня экспрессии гена микродистрофина с исходным с помощью иммунофлуоресценции и вестерн-блоттинга [32].
В 2022 году было проведено еще одно рандомизированное двойное слепое плацебо-контролируемое исследование SRP-9001 путем измерения биологических и клинических конечных точек в трех частях: два 48-недельных рандомизированных, двойных слепых, плацебо-контролируемых периода и открытый период наблюдения (часть 3). Пациенты, рандомизированные в группу плацебо в части 1, получили возможность лечения с помощью SRP-9001 в части 2. В исследовании приняли участие дети от 3 до 6 лет включительно, с установленным клиническим диагнозом «мышечная дистрофия Дюшенна».
Первая часть вышеупомянутого исследования была направлена на отслеживание изменений уровня микродистрофинов в организме и показателей NSAA. Согласно полученным результатам, пациенты, получавшие SRP-9001, достигли средней активности микродистрофина на уровне 28,1% после 12 недель лечения. Кроме того, показатели NSAA у мальчиков в возрасте от 4 до 5 лет, получавших SRP-9001, значительно улучшились по сравнению с теми, кто получал плацебо.
Вторая часть исследования была посвящена оценке пациентов, которые в первой части получали плацебо, но затем им был назначен SRP-9001. Сравнение показателей у этих пациентов и контрольной группы, сопоставимой по возрасту и использованию стероидов, продемонстрировало, что показатель NSAA у пациентов, получавших SRP-9001, был в среднем на 2 балла выше. В частности, группа, получавшая SRP-9001, увеличила свой показатель NSAA на 1,3 балла, в то время как в контрольной группе он снизился на 0,7 балла.
Для анализа использовался индекс NSAA, демонстрирующий динамику общего балла амбулаторной оценки North Star, измеряемый относительно начальных показателей. В исследовании анализировались параметры, второстепенные по своему значению: изменения в способности испытуемых подниматься с земли, время преодоления дистанций в 10 и 100 метров. Дополнительно изучалось варьирование уровней экспрессии микродистрофина от отмеченной базовой точки, количественно определяемое по проценту позитивно окрашенных волокон дистрофина (IF Percent Dystrophin Positive Fibers) и с применением метода иммунной флуоресценции [31].
Заключение
Анализ литературы и клинического исследования продемонстрировал, что SPR-9001 обеспечивает функциональное улучшение, которое превосходит наблюдаемое при стандартном лечении, хорошо переносится и вызывает минимальные побочные эффекты.
Библиографическая ссылка
Сафина С.А., Матвеева К.А., Валиуллина З.А., Старцева Л.В., Богданова Ю.А., Сулейманова Г.Ф., Самородов А.В. ГЕННАЯ ТЕРАПИЯ АДЕНОАССОЦИИРОВАННЫМИ ВИРУСАМИ В ЛЕЧЕНИИ МЫШЕЧНОЙ ДИСТРОФИИ ДЮШЕННА // Современные проблемы науки и образования. 2024. № 6. ;URL: https://science-education.ru/ru/article/view?id=33751 (дата обращения: 29.06.2026).
DOI: https://doi.org/10.17513/spno.33751