



Первичная цилиарная дискинезия (ПЦД) – это редкое наследственное заболевание, которое характеризуется поражением ресничек на поверхности дыхательных путей и подобных им структур, что приводит к нарушению их двигательной активности. Заболевание встречается с частотой от 1:10000 до 1:40000 новорожденных [1]. Основные симптомы включают поражение всех отделов дыхательной системы, что приводит к формированию хронического воспаления и бронхоэктазов. У пациентов с ПЦД часто наблюдаются бесплодие у мужчин и эктопические беременности у женщин. Приблизительно у половины пациентов с ПЦД выявляется полное или частичное обратное расположение внутренних органов [2]. На сегодняшний день известно более 30 генетических локусов, ответственных за развитие данной патологии. Помимо более распространенных мутаций, у пациентов с ПЦД могут быть выявлены и редкие генетические дефекты [3].
Диагностика ПЦД часто занимает длительное время из-за недостаточности знаний о заболевании у врачей и сложности проведения или недоступности специализированных исследований. Это приводит к задержке осуществления необходимого лечения пациентов и быстрому снижению легочной функции. Терапия ПЦД направлена на улучшение дренажной функции бронхов и купирование воспалительных изменений (назначение антибактериальных препаратов при обострении бронхолегочного процесса) [4, 5]. Учитывая, что на сегодняшний день отсутствует этиотропная терапия заболевания, направленная на коррекцию генетических дефектов, раннее выявление заболевания и начало специализированного медицинского наблюдения пациентов с ПЦД выходят на первый план.
Цель исследования: описать клиническое наблюдение трех сибсов с различными вариантами ПЦД, подтвержденными наличием генетических нарушений в гене DNAH11, что проявлялось наличием и отсутствием такого симптомокомплекса, как транспозиция внутренних органов (лат. situs inversus).
Материал и методы исследования
Проведен анализ историй болезни пациентов, представлены результаты световой микроскопии биоптата слизистой из полости носа (для оценки функциональной активности реснитчатого эпителия) и результаты молекулярно-генетической диагностики.
Результаты исследования и их обсуждение. Представлен семейный случай, выявленный и наблюдаемый в Чувашской Республике (ЧР). В одной семье из пятерых детей у троих диагностирована первичная цилиарная дискинезия: у брата синдром Картагенера, у двух сестер классический вариант заболевания.
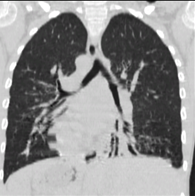
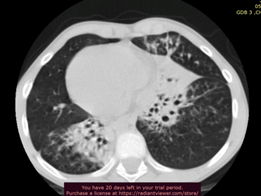
Первый обследованный, больной Д., впервые был доставлен на обследование в БУ «Республиканская детская клиническая больница» МЗ Чувашии возрасте 6 лет (2008 г.) с жалобами на длительный влажный кашель с трудноотделяемой мокротой, одышку, заложенность носа, повышение температуры тела до 39ºС, слабость, потливость, быструю утомляемость. По данным сбора анамнеза известно, что ребенок рожден в Республике Армения, а на территории ЧР семья проживает с 2006 г. Беременность третья, роды третьи, срочные. При рождении имел массу 2500 г, длину 46 см. Родители здоровы, в кровном родстве не состоят. У старшей сестры – частые бронхиты, риносинуситы, у двоюродного брата по материнской линии – нарушение слуха, у сына двоюродного брата по материнской линии – врожденный порок сердца, частые бронхиты. Мальчик болен с рождения, перенес респираторный дистресс-синдром, двустороннюю пневмонию. Со слов родителей, с рождения постоянный кашель, заложенность носа, периодические подъемы температуры тела до фебрильных цифр. Ребенок неоднократно переносит синуситы, отиты, бронхиты, пневмонии, выслушиваются постоянные влажные хрипы в легких. В 4 года проведена аденотонзиллотомия (в Республике Армения), при подготовке к которой родителям впервые сообщили о транспозиции внутренних органов ребенка. При осмотре состояние тяжелое. Затрудненное носовое дыхание. Одышка при физической нагрузке. Сатурация кислорода – 97%. Ребенок со сколиотической осанкой, плоско-вальгусной деформацией стоп. В легких с обеих сторон выслушивались множественные разнокалиберные влажные хрипы. В ходе обследования у ребенка был проведен дифференциальный диагноз и исключен муковисцидоз (потовый тест на аппарате «Нанодакт» показал значение 32 ммоль/л, что соответствует нормальному значению). Также обнаружены двусторонняя пневмония, утолщение бронхов и признаки мукостаза, уменьшение средней доли слева за счет частичного фиброателектаза; диагностированы двусторонний гайморит, этмоидит; микробиологическое исследование промывных вод бронхов выявило высев синегнойной палочки. С учетом данных анамнеза, обследования пациенту был выставлен диагноз: «Врожденный порок развития бронхолегочной системы: синдром Картагенера. Хронический бронхит. Хронический риносинусит. Фиброателектаз средней доли слева». Ребенок постоянно наблюдался и лечился в клинике по месту жительства: наблюдение пульмонолога, оториноларинголога; 1–2 раза в год госпитализировался в стационар. Антибактериальная терапия проводилась с учетом чувствительности флоры. Постоянно проводились кинезиотерапия, ингаляционная муколитическая и бронхолитическая терапия. Несмотря на проводимую терапию, положительная динамика отсутствовала. Сохранялся постоянный влажный кашель с трудноотделяемой гнойной мокротой, периодами до рвоты; беспокоили обострения бронхолегочного процесса 4–5 раз в год в виде пневмонии; с 10 лет присоединилась одышка в покое, усиливающаяся при физической нагрузке; прогрессировали рентгенологические изменения – с 10 лет двусторонние цилиндрические бронхоэктазы, пневмофиброзные изменения (рис. 1, 2); показатели спирометрии – выраженные смешанные нарушения; отмечено прогрессирующее снижение слуха, в 12 лет диагностирована двусторонняя кондуктивная тугоухость 1–2-й степени; сохранялось длительное затруднение носового дыхания с периодическими обострениями хронического риносинусита, формированием хронического евстахеита, хронического экссудативного среднего отита. В посевах мокроты
 |
 |
определялся рост штаммов Ps. aeruginosa.


Нарушение осанки прогрессировало, в 12 лет установлен диагноз «диспластический S-образный сколиоз грудопоясничного отдела позвоночника 1 ст.», «килевидная деформация грудной клетки». Ребенка беспокоили симптомы интоксикации (слабость, снижение аппетита, потливость), отмечалось отставание в физическом развитии. В возрасте 13 лет пациент обследован в условиях НИКИ педиатрии и детской хирургии имени академика Ю.Е. Вельтищева. Диагноз ПЦД был подтвержден, результаты биоптата слизистой оболочки носа свидетельствовали об отсутствии движения ресничек мерцательного эпителия. Уровень оксида азота в выдыхаемом воздухе (0,4 ppd) соответствовал сниженному уровню, характерному для ПЦД. Полученные данные комплексного обследования указывают на наличие у ребенка классического синдрома Картагенера, который является формой ПЦД, характеризующейся комбинацией респираторных симптомов, инверсией внутренних органов, поражением легких в виде нарушения мукоцилиарного клиренса и формирования бронхоэктазов, хроническим синуситом.
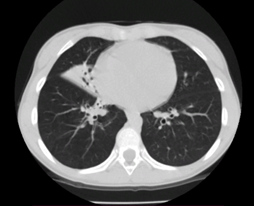
Вторая пациентка З., старшая сестра пациента Д., впервые поступила в клинику на обследование в возрасте 8 лет. Ребенок от второй беременности, от вторых срочных родов. Масса при рождении 2700 г, длина 49 см. В роддоме у девочки диагностировали двустороннюю пневмонию. Жалобами при поступлении были длительный влажный кашель, персистирующая заложенность носа, повторные пневмонии, респираторные заболевания, которые всегда осложнялись бронхитами. При поступлении физическое развитие оценивалось как среднее, гармоничное. Одышки в покое не было, насыщение крови кислородом (SatО2) составляло 99%. При осмотре выявлены заложенность носа и обильные влажные хрипы в легких с обеих сторон. По данным рентгенологического исследования– двусторонняя пневмония, двусторонний гайморит, этмоидит. Спирометрия – смешанные нарушения. Выполнен потовый тест – 21 ммоль/л (норма — до 60 ммоль/л). Микробиологический анализ мокроты на микрофлору выявил рост гемолитического стрептококка. С диагнозами «Хронический бронхит. Хронический риносинусит» ребенок находился на учете у пульмонолога, ЛОР врача по месту жительства. Постоянный влажный кашель и заложенность носа, обострения бронхита 3–4 раза в год свидетельствовали об отсутствии эффективности получаемой терапии (курсы муколитической терапии, по показаниям антибактериальная и бронхолитическая терапия) и прогрессировании респираторных симптомов в динамике. Данные динамического исследования КТ ОГК также свидетельствовали о прогрессирование хронического воспаления дыхательной системы у ребенка. В 11 лет были выявлены цилиндрические бронхоэктазы правого легкого, а в 13 лет – фиброателектаз средней доли, частичный фиброателектаз С7 правого легкого, явления диффузного бронхиолита в нижних долях легких, более выраженно справа, мукостаза (рис.
 |
3).
Рис. 3. Компьютерная томограмма пациентки З., 13 лет
По данным спирометрии – смешанные нарушения с нарастанием обструктивных нарушений. В мокроте по данным бакпосевов высевались бета-гемолитический стрептококк и золотистый стафилококк. При проведении аудиометрии в 13 лет установлена двусторонняя кондуктивная тугоухость 1-й степени. Ребенок наблюдался у ортопеда с диагнозом «Сколиотическая осанка. Продольное плоскостопие 2 ст.». Однократно перенесла острый вторичный пиелонефрит. Учитывая данные анамнеза, пациентка вместе с братом направлена в НИКИ педиатрии и детской хирургии имени академика Ю.Е. Вельтищева. Диагноз при направлении: «Хронический бронхит. ДН 2 ст. Хронический риносинусит. Двусторонняя кондуктивная тугоухость 1 ст.». Проведенное обследование, включающее биопсию слизистой носа (отсутствие двигательной активности ресничек респираторного тракта), уровень оксида азота в выдыхаемом воздухе (–0,5 ppd, показатель ниже нормы) позволили установить диагноз ПЦД.
Младшая сестра впервые поступила на обследование в клинику в возрасте 4,5 года. Основные жалобы при поступлении – на постоянный продуктивный кашель с отхождением небольшого количества слизисто-гнойной мокроты, постоянную заложенность носа с обильным отделяемым, а также периодические подъемы температуры тела до фебрильных цифр. Ребенок от 6-й беременности, от 5-х срочных родов. Оценка по Апгар 6/9 баллов, масса при рождении 3300 г, длина тела 52 см. Больна с рождения, диагностированы синдром дыхательных расстройств, ранняя неонатальная правосторонняя пневмония. С первых месяцев жизни рецидивирующие обструктивные бронхиты (до 4 раз в год), постоянная заложенность носа, кашель с трудноотделяемой мокротой. С 3 лет наблюдается у ЛОР врача с хроническим назофарингитом, неоднократно переносит двусторонний острый гнойный гайморит. При осмотре: физическое развитие среднее, гармоничное, одышки в покое нет, сатурация кислорода (SatО2) – 96%, в легких выслушиваются влажные хрипы. Потовая проба отрицательная –30 ммоль/л эквNaCl («Нанодакт»). Выполнена КТ ОГК, выявившая КТ-признаки пневмофиброза SV правого легкого. В анализе мокроты на микрофлору клинически значимые аэробные, факультативно-анаэробные микроорганизмы не обнаружены. Оценка двигательной активности ресничек эпителия слизистой оболочки носа проведена трижды (НИКИ педиатрии и детской хирургии имени академика Ю.Е. Вельтищева). В возрасте 5 лет убедительных данных за ПЦД не получено. Диагноз ПЦД установлен в возрасте 7 лет (выявлены снижение двигательной активности мерцательного эпителия, снижение уровня оксида азота в выдыхаемом воздухе –0,4 ppd (N 10-20 ppd)). В возрасте 9 лет пациентке проведено молекулярно-генетическое исследование. По результатам проведенного исследования выявлены 2 гетерозиготные мутации в гене DNAH11, отвечающие за развитие ПЦД, с/без обратного расположения внутренних органов (situs inversus), тип 7 (OMIM 611884).
В динамике отмечается прогрессирующее ухудшение состояния пациентки. Болезнь проявляется постоянным влажным кашлем с отхождением вязкой слизисто-гнойной мокроты, стойкой выраженной заложенностью носа, одышкой при физической нагрузке, постоянно выслушивающимися при аускультации сухими и влажными хрипами. Обострения бронхолегочного процесса 5–6 раза в год, затяжные, с гипертермией и нарастанием количества гнойной мокроты. В возрасте 5 лет проведена аденотонзиллотомия, с 8 лет диагностирована двусторонняя дисфункция слуховых труб. По результатам посевов мокроты преобладает наличие Staphylococcus аureus. Показатели спирометрии указывают на резко выраженные смешанные нарушения с признаками значительной неравномерности вентиляции и обструктивных нарушений. КТ органов грудной клетки и придаточных пазух носа (рис. 4) показывает снижение пневматизации обеих гайморовых пазух, искривление носовой перегородки, признаки выраженного обструктивного бронхита, бронхиолита с отрицательной динамикой, субтотального фиброателектаза средней доли, пневмофиброзных изменений легких поствоспалительного характера.
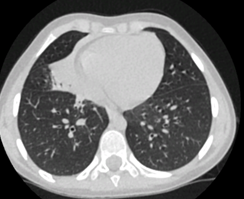 |
Рис. 4. Компьютерная томограмма пациентки В., 9 лет
Таким образом, представленный клинический случай, демонстрирующий диагностику различных вариантов ПЦД (с и без наличия транспозиции внутренних органов) у двух сибсов, подтверждает аутосомно-рецессивный путь наследования синдрома ПЦД, а также возможную фенотипическую гетерогенность у пациентов с одинаковым патогенным вариантом генотипа. Действительно, в настоящее время нет единого алгоритма диагностики ПЦД. Для установления диагноза учитывается комплексный подход, включающий характерную клиническую картину, результаты специализированных исследований и анализов [4, 5]. В представленном клиническом случае окончательный диагноз пациентам был установлен на основании анамнеза, жалоб, клинических проявлений, а также результатов анализа уровня оксида азота (NO) в выдыхаемом воздухе и анализа частоты и паттерна биения ресничек в биоптате из полости носа с помощью световой микроскопии. Морфометрическое исследование выявило резко выраженные и значительные нарушения цилиарной функции эпителия. В описанном примере характерная клиническая симптоматика манифестировала у наблюдаемых сибсов уже с периода новорожденности. В последующем отмечено прогрессирование заболевания с формированием хронического риносинусита, хронического бронхита. Среди описанных сибсов наиболее тяжелое течение заболевания описано у пациента с синдромом Картагенера как совокупность проявлений в виде раннего формирования бронхоэктазов, развития тугоухости, хронического высева синегнойной палочки, непрерывно-рецидивирующего течения бронхолегочного процесса, приводящего к снижению функции внешнего дыхания, синдрому хронической интоксикации и снижению физического развития. Младшему ребенку проведен молекулярно-генетический анализ, в результате которого диагноз ПЦД полностью подтвержден. Генетическая основа разнообразных дефектов, влияющих на структуру и функцию ресничек при ПЦД, не ясна: на сегодняшний день упомянуты мутации примерно в 200 генах, приводящие к ультраструктурным изменениям ресничек мерцательного эпителия трахеобронхиального дерева и потере их нормальной функции, наследующихся преимущественно аутосомно-рецессивно [6, 7, 8]. Заболевание возникает как прямой результат врожденных дефектов подвижных ресничек, покрывающих респираторный эпителий, что приводит к нарушению мукоцилиарного клиренса. При секвенировании экзома у наблюдаемой пациентки был определен ранее не описанный вариант нуклеотидной последовательности в экзоне 36 гена DNAH11 (rs372143147) в гетерозиготном состоянии, который приводит к приобретению преждевременного стоп-кодона. Вариант встречается в базе данных популяционных частот gnomADv3.1 с низкой частотой, с большой вероятностью приводит к потере соответствующей копии гена. Также в экзоне 63 гена DNAH11обнаружен ранее описанный вариант нуклеотидной последовательности (rs764509824) в гетерозиготном состоянии, вызывающий аминокислотную замену p.Gly3422Arg. Вариант не встречается в базе данных популяционных частот gnomADv3.1, располагается в консервативном сайте и предсказан оказывать патогенный эффект компьютерными алгоритмами. Патогенные биаллельные варианты в гене DNAH11 приводят к развитию ПЦД как с, так и без обратного расположения внутренних органов, тип 7 (OMIM № 611884). Известно, что мутации в гене DNAH11 приводят к изменению структуры тяжелых цепей динеиновых ручекаксонемы [9]. Ультраструктура цилиарной системы у пациентов с мутациями в генеDNAH11является нормальной.Мутации DNAH11могут вызывать дисфункцию цилиарной системы, изменяя движение ресничек, что приводит к ухудшению мукоцилиарного клиренса. Таким образом, представлен клинический случай, в котором были продемонстрированы разные варианты ПЦД в одной семье, что дополнительно подтверждает способность мутации в гене DNAH11 приводить к фенотипической гетерогенности.
Заключение. Продемонстрировано развитие различных вариантов ПЦД среди трех сиблингов, а также наличие здоровых детей в семье. Ознакомление врачей с клиническими вариантами течения болезни будет способствовать повышению настороженности врачей в отношении хронического респираторного синдрома и лучшему выявлению пациентов, нуждающихся в углубленном обследовании.
Библиографическая ссылка
Голубцова О.И., Краснов М.В., Будылина М.В., Сергиенко Д.Ф., Иванова Т.Ю., Скворцова И.А., Андреев А.В. СЕМЕЙНЫЙ СЛУЧАЙ ПЕРВИЧНОЙ ЦИЛИАРНОЙ ДИСКИНЕЗИИ // Современные проблемы науки и образования. 2024. № 3. ;URL: https://science-education.ru/ru/article/view?id=33510 (дата обращения: 14.07.2026).
DOI: https://doi.org/10.17513/spno.33510