



Врожденные деформации позвоночника (ВДП) представляются одним из сложных и актуальных разделов современной ортопедии. Причиной врожденных деформаций позвоночного столба являются аномалии развития тел позвонков [1]. Несмотря на то что частота встречаемости врожденных пороков развития позвоночного столба сравнительно невелика (0,5–1 на 1000 новорожденных), они могут приводить к тяжелым и ригидным искривлениям позвоночника уже в дошкольном возрасте, а также к необратимым нарушениям со стороны внутренних органов, прежде всего со стороны дыхательной и сердечно-сосудистой систем [2]. Отмечено, что врожденные деформации позвоночника чаще встречаются у девочек по сравнению с мальчиками с соотношением 2,5:1 [3].
Искривления позвоночного столба значительно влияют на физическое и психологическое состояние пациентов. Кроме того, врожденные деформации позвоночника являются одной из причин формирования инвалидности среди детского населения. Согласно литературным данным, установлено, что около 50% врожденных сколиозов может сочетаться с пороками и дисфункцией других органов [4], включая заболевания почек, сердца и интраспинальные пороки.
Тесная эмбриологическая связь между позвоночником и спинным мозгом может привести к сопутствующим внутриспинальным аномалиям, таким как расщепление спинного мозга (SCM), фиксированный спинной мозг, низкорасположенный конус, сирингомиелия и интраспинальные новообразования. Врожденные деформации позвоночника относительно ригидны, могут быстро прогрессировать и, когда они связаны с аномалиями позвоночного канала, имеют более высокий риск неврологических осложнений. Будучи эмбриологической аномалией, ВДП часто сочетается с пороками развития мочеполовой, сердечно-сосудистой, скелетно-мышечной систем. Эти факторы играют важную роль при принятии решений о тактике лечения детей с ВДП [5].
Цель исследования
В связи с вышеизложенным целью нашего обзора явилось последовательное описание процессов формирования позвоночника в эмбриогенезе и в постнатальном периоде, а также акцентирование внимания на выявлении генетических и средовых факторах, которые могут привести к появлению врожденных деформаций позвоночника. Мы считаем чрезвычайно важным, особенно в отношении практикующих хирургов-ортопедов, выявить те генетические факторы, которые могут привести к быстрому прогрессированию ВДП и способны служить руководством для принятия решения о назначении хирургического лечения при выявлении такого набора прогностических факторов.
Этиология врожденных патологий позвоночника
Процесс сомитогенеза контролируется генами сигнальных путей Notch, WNT и FGF. Они координируют ключевые этапы сомитогенеза, включая сегментацию, двустороннюю симметрию и формирование позвонков. Нарушение сигнальных путей и сбой регуляторов этих процессов имеют большое значение в патогенезе врожденных деформаций позвоночника. Развитие позвонков происходит за счет синхронной конвергенции нескольких проводящих путей и нескольких десятков генов. Выявлено, что гены сигнальных путей Notch, WNT и FGF могут мутировать, и эти мутации выявляются у пациентов с ВДП. Путем использования различных методологических подходов было идентифицировано несколько наиболее важных генов-кандидатов (рис. 1).
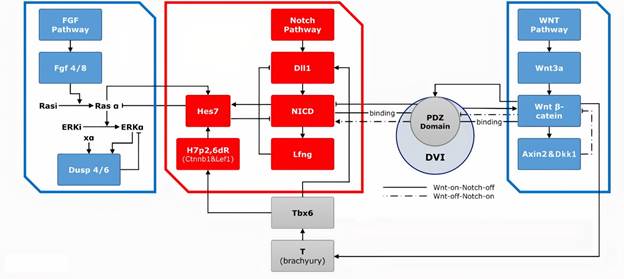
Рис. 1. На рисунке отражены три основных пути взаимодействующих генов и эффекторов в сомитогенезе. Показаны компоненты часов сегментации (красная рамка) и компоненты волнового фронта (синяя рамка). Часы сегментации в основном состоят из пути Notch и ряда генов и эффекторов. Dll1 и нисходящие NICD и Lfng периодически экспрессируются через цикл обратной связи, формируя модель «часов». Градиенты сигналов FGF и WNT составляют волновой фронт [6]
В сигнальном пути Wnt Wnt3a индуцирует экспрессию нижележащих P-cat и Axin2. Белки AXIN2 и DKK1 являются ингибиторами P-cat с отрицательной обратной связью, что способствует формированию регуляторного цикла. Wnt3a индуцирует экспрессию Tbx6, который дополнительно активирует экспрессию Dll1 и связан с NICD через Hes7, тем самым устанавливая коммуникативную связь сигнальных путей Notch и WNT. В сигнальном пути FGF гены Fgf4 и Ff8 индуцируют экспрессию pERK, что дополнительно вызывает экспрессию нижележащих Dusp4 и Dusp6. Сигнальная осцилляция FGF также основана на механизме обратной связи [6].
Мутации в генах семейств транскрипционных факторов HES и MESP приводят к врожденному искривлению позвоночника [7], что указывает на их важность в развитии сомитов. Гены MESP кодируют факторы транскрипции семейства bHLH, а делеция MESP2 у мышей нарушает сегментацию и рострокаудальную полярность сомитов [8]. У рыбок данио четыре гомолога MESP2 (mespaa, mespab, mespba и mespbb) динамически экспрессируются в передней пресомитной мезодерме (PSM) с пиками, которые совпадают со впадинами в паттернах экспрессии часовых генов her1 и her7 и гена deltaC, кодирующего лиганд сигнального пути Notch [9]. У рыбок данио репрессор транскрипции, кодируемый RIPPLY1, экспрессируется в отдельных участках в передней PSM и во вновь образованных сомитах [10]. Опосредованный морфолино-олигонуклеотидом нокдаун RIPPLY1 у рыбок данио или нокаут RIPPLY2 у мышей вызывают дефекты сегментации [10, 11]. Мутации в MESP2 и RIPPLY2 присутствуют у пациентов с врожденным сколиозом [12, 7] или синдромом Клиппеля–Фейля [13] соответственно. Мутации в генах Delta, MESP и HES приводят к сколиозу [14 15], а мутации их ортологичных генов в модели на мышах или рыбках данио полностью (частично) воспроизводят фенотип [15, 16]. Хотя функциональное значение этих генов было установлено на мышиной модели, динамические регуляторные отношения между этими генами еще не идентифицированы ни в одном модельном организме.
Помимо мутаций в отдельных генах, приводящих к врожденным деформациям позвоночника, было исследовано множество случаев, когда ген, непосредственно вызывающий врожденное искривление позвоночника в результате хромосомных мутаций (de novo – различных транслокаций, инверсий и т.д.), образовывал группу сцепления с другими генами, что дополнительно приводило к сопутствующим порокам развития других органов [17]. Таким образом, традиционный анализ сцепления имеет значительные ограничения для идентификации генов-кандидатов, поскольку ненаследственные типы врожденного сколиоза часто являются спорадическим случаем, который может зависеть от мутаций, вызванных различными тератогенными факторами во время беременности.
Другие гены-кандидаты врожденных деформаций позвоночника также были идентифицированы на животных моделях. В фенотипически однородной когорте пациентов с этой патологией для анализа были отобраны пять генов-кандидатов: PAX1 и T(Brachyury) [18, 19], а также WNT3A, DLL3 и SLC35A3 [20–22], экстраполированных из мышиных моделей. Области локализации ВДП, представленные в этой когорте, охватывали всю длину позвоночника и были описаны более подробно в предыдущем сообщении [18]. У пациентов с врожденным сколиозом варианты последовательности PAX1, DLL3, WNT3A и T (Brachyury), связанные со снижением пенетрантности, были идентифицированы и наблюдались с низкой частотой или не обнаруживались у здоровых людей [19]. Когорта из 79 случаев врожденного сколиоза была исследована на наличие вариантов в генах DLL3, MESP2 и HES7 (гены, связанные с ВДП и спондилокостальным дизостозом). Одна семья имела мутации в MESP2, другая – в HES7. В обеих семьях пенетрантность и выраженность мутаций были разными; также было показано, что они нарушают in vitro функцию факторов транскрипции, кодируемых этими генами [23]. В другой когорте, состоящей из 154 пациентов с врожденным сколиозом, было обнаружено, что ген LMX1A связан с чувствительностью к этой патологии у этнических китайцев [24].
Биаллельные мутации гена CDK10 (циклин-зависимая киназа 10) зародышевой линии были идентифицированы в пяти родственных семьях Саудовской Аравии с задержкой роста, слиянием позвонков или образованием полупозвонков и задержкой в развитии. CDK10 – протеинкиназа, которая играет регуляторную роль в транскрипции [25]. Мыши с нокаутом CDK10 имели несколько костных дефектов, влияющих на осевой скелет, такие дефекты могут проявляться у людей с мутациями CDK10 [25]. Рецессивные миссенс-мутации были обнаружены в гене SLC35A3, который характеризуется комплексом врожденных дефектов [26]. К дефектам позвоночника относились бабочковидные позвонки и полупозвонки, распределенные по всему позвоночнику, а также «волчья пасть», дефекты сердечной перегородки и сосудов, укороченные конечности и лицевой дисморфизм [27]. Также был разработан метод хромосомного микроматричного анализа (CMA) для обнаружения изменений в ДНК по всему геному. Преимущество CMA заключается в возможности идентифицировать потенциальные области микроанеуплоидии, связанные с врожденным сколиозом (ВС) по всему геному, а не ограничиваться только одной небольшой областью. Поскольку этиология ВС неоднородна и может включать несколько генетических дефектов, многие из которых еще предстоит определить, этот подход обеспечивает эффективный инструмент скрининга для выявления дополнительных локусов, которые могут маскировать генетические дефекты, лежащие в основе этиологии ВДП. С помощью CMA успешно выявили некоторые потенциальные области, связанные с ВС, в частности с идентификацией области делеции гена TBX6, что определяет значительный прогресс в нашем понимании причин ВС [28].
В клинической практике у определенной части пациентов с ВС не наблюдается заметных дополнительных органических деформаций. Эти случаи вызывают большой интерес для геномных исследований, потому что они представляют собой фенотипически отличную группу для изучения патогенеза деформаций позвоночника. Варианты генов, участвующих в сомитогенезе, были изучены Ghebranious et al. [19], когда группа пациентов с гетерогенными типами ВС была секвенирована с использованием панели генов, связанных с сигнальными путями в сомитогенезе, включая PAX1, DLL3, SLC35A3, WNT3A, TBX6 и T (Brachyury). Были просеквенированы весь T (Brachyury), а также кодирующие области, сайты сплайсинга и 500 п.н. промоторной области TBX6. У трех неродственных пациентов была такая же c.1013C>T транзиция в экзоне 8 гена T, но не было выявлено никаких полиморфных последовательностей в гене TBX6. Fei et al. [29] генотипировали два известных SNP в гене TBX6 среди 254 этнических китайцев (127 пациентов с ВС и 127 в контроле). При анализе аллелей SNP rs2289292 (SNP1, chr16: 30005131, G/A, экзон 8) и rs3809624 (SNP2, chr16: 30010303, A/G, 5'-нетранслируемая область) частоты значительно различались между пациентами и контрольной группой (P=0,017 и 0,033 соответственно). Анализ гаплотипов показал значительную взаимосвязь между SNP1/SNP2 при ВС (P=0,017) с гаплотипом G-A, который чаще наблюдается в контроле (отношение шансов 0,71; 95%-ный доверительный интервал 0,51–0,99).
Ген TBX6 известен как член Т-box семейства и кодирует фактор транскрипции, который играет важную роль в регуляции процессов развития. TBX6 расположен на хромосоме 16p11.2, размер 6091 п.н. и содержит 9 экзонов. Сообщалось, что взаимодействия между TBX6 и генами, участвующими в модели волнового фронта, или самим TBX6 приводят к аномальному образованию сомитов, способствующих ВС [30]. Несколько исследований показали, что вариация числа копий (CNV) в области 16p11.2 может быть связана с фенотипом ВС. Shimojima et al. [31] сообщили о трехлетнем мальчике с задержкой в развитии: паховая грыжа; полупозвонок Т10, Т12 и L3; отсутствует правое 12-е ребро; гипоплазия левого 12-го ребра. У пациента была делеция 593 КБ в области 16p11.2, а у матери была такая же делеция, что было выявлено с помощью CMA-анализа. Al-Kateb et al. [32] проанализировали радиологические данные, полученные от 10 пациентов с CNV области 16p11.2 (9 с делециями и 1 с дупликацией). У 8 из них был ВС, а у остальных 2 – идиопатический сколиоз (ИС). Также были обследованы 5 пациентов с ранее выявленными изменениями в области 16p11.2. и сходными скелетными аномалиями, у 2 был ВС, у остальных – ИС.
Хотя во многих исследованиях сообщается о связи между 16p11.2 CNV и ВС, точный механизм здесь до сих пор не ясен. Впоследствии Wu et al. [28] обнаружили, что нулевые варианты TBX6 и Tbx6 общего гипоморфного аллеля вместе вносят вклад в развитие ВС, находясь в компаудном состоянии. В группе из 161 китайского пациента со спорадическим неродственным ВС анализ CNV выявил 17 гетерозиготных нулевых мутаций TBX6 у этих индивидуумов с ВС. Это включало 12 случаев рекуррентной делеции 16p11.2, затрагивающей TBX6, и 5 однонуклеотидных вариантов (1 нонсенс-мутация и 4 мутации со сдвигом рамки считывания). Мутаций TBX6 в контрольной группе не было обнаружено. Идентификация фенотипически нормальных индивидуумов с микроделециями 16p11.2 и дискордантными внутрисемейными фенотипами ВС у носителей микроделеций 16p11.2 показала, что гетерозиготной нулевой мутации TBX6 недостаточно, чтобы вызвать ВС. Примечательно, что был идентифицирован другой распространенный (около 44% в азиатской и 33% в европейской популяциях) гаплотип, который, как было показано, является гипоморфным аллелем в сочетании с нулевыми мутациями TBX6. Это компаудное наследование редких и распространенных (CIRC) паттернов составляло до 11% спорадических случаев ВС. Эти результаты были подтверждены в дополнительной когортной и многоцентровой серии исследований микроделеций 16p11.2 и в дальнейшем подтверждены в исследованиях в японской когорте ВС [33] и французской когорте SDV (дефекты сегментации позвонков) [34].
Кроме того, недавно Feng et al. [35] обнаружили, что 2 пациента с полупозвонком в грудном отделе позвоночника и 1 пациент с полупозвонком в поясничном отделе позвоночника являлись носителями ранее идентифицированного гетерозиготного варианта патогенного компаунда TBX6. Кроме того, полноэкзомное секвенирование пациентов с ВС и их семей выявило миссенс-мутацию de novo (c.G47T: p.R16L) в другом гене семейства T-box – TBXT. Эта редкая мутация нарушает связывание TBXT с его последовательностью-мишенью, что приводит к снижению транскрипционной активности и оказывает доминирующее негативное влияние на TBXT дикого типа.
Liu J. et al. [36] определили новый подтип ВС, названный TACS, содержащий компаунд TBX6 LoF с гипоморфным аллелем. TACS – это нозологическая единица, определяемая последовательными клинически измеряемыми эндофенотипами, т.е. более молодой возраст в начале заболевания, полупозвонки/бабочковидные позвонки, затрагивающие нижнюю часть позвоночника, простая реберная аномалия и меньшее количество позвоночных и интраспинальных дефектов. TACScore может определять и направлять клиническое ведение, а также генетическое и клиническое геномное тестирование. Исследования на людях и мышах дополнительно подтверждают модель компаудного наследования и дозы генов, а также дают представление о потенциальных биологических последствиях нарушений дозы и экспрессии гена TBX6/Tbx6 для развития позвоночника. Такие генетические модели могут иметь значение и для других врожденных дефектов.
Взаимодействие генов сомитогенеза и тератогенных факторов окружающей среды
Помимо генетической этиологии, влияние вредных факторов окружающей среды на развивающийся эмбрион может вызывать определенные дефекты, в том числе и в позвоночнике. Например, имеется достаточно эпидемиологических данных о том, что факторы окружающей среды, такие как курение матери или диабет, увеличивают риск врожденных дефектов [37, 38]. Однако, поскольку образование сомитов у людей происходит между 3-й и 5-й неделями, трудно доказать, что дефекты позвоночника, выявляемые при рождении, являются результатом тератогенного воздействия окружающей среды в то время, когда мать, возможно, не знала о своей беременности.
Таким образом, анализ большинства факторов риска окружающей среды, способствующих возникновению дефектов позвоночника, был ограничен модельными системами на животных. Действительно, подобные эксперименты на животных проводятся уже много лет. Эти эксперименты представили доказательства того, что токсически действуют такие разнообразные факторы, как ретиноевая кислота, вальпроевая кислота, диабет матери, гипоксия плода, окись углерода, мышьяк, этанол, гипертермия, дефицит цинка у матери, фосфорорганические пестициды, ингибирование выработки оксида азота и борной кислоты, и другие факторы, являющиеся потенциальными тератогенами окружающей среды [39, 40]. Конечно, ни генетические факторы, ни факторы окружающей среды, как правило, не действуют изолированно, а взаимодействие этих факторов (гена и окружающей среды) будет влиять на пенетрантность и выраженность причин, вызывающих дефекты позвоночника.
Эта гипотеза подтверждается использованием близнецового метода. Например, в большинстве описанных случаев монозиготных близнецов с врожденным сколиозом был болен только один близнец. Если поражены оба близнеца, локализация и тяжесть дефектов позвоночника различаются. Таким образом, Sparrow et al. предоставили первое экспериментальное свидетельство в поддержку этой гипотезы [23]. Были обследованы две семьи, в которых наблюдался небольшой дефект позвоночника. В родословных этих двух семей с врожденным сколиозом были выявлены мутации в генах MESP2 и HES7. Секвенирование генов-кандидатов показало, что эти семьи действительно несут мутантные аллели MESP2 и HES7 соответственно. В обеих семьях все пациенты с ВС были гетерозиготны по мутантному аллелю, и наоборот, не все гетерозиготы имели дефекты позвоночника. Также были исследованы нарушения скелета линий мышей, несущих нулевые аллели MESP2 и HES7.
Приблизительно 50% гетерозиготных эмбрионов мышей HES7 и 10% гетерозиготных эмбрионов MESP2 имели дефекты позвонков. Таким образом, как у людей, так и у мышей индивидуумы, гетерозиготные по делеционным мутациям в генах MESP2 и HES7 пути Notch, имеют врожденный сколиоз с низкой пенетрантностью. Предполагалось, что на пенетрантность и выраженность врожденного сколиоза влияет окружающая среда, и в качестве потенциального фактора окружающей среды была выбрана острая гестационная эмбриональная гипоксия. Исследования, начатые еще в XIX в., показали, что снижение уровня кислорода, необходимого для эмбрионов позвоночных, может вызывать грубые структурные аномалии, включая дефекты позвонков, очень похожие на те, которые наблюдаются у гетерозиготных мышей HES7. Это относится и к беременности у человека, поскольку внутриутробная гипоксия может быть вызвана многими факторами окружающей среды. Было продемонстрировано, что воздействие экстремальной гипоксии матери (5,5% кислорода) в течение 8 ч во время формирования тканей-предшественников позвоночника (сомитов) вызывает серьезные дефекты позвоночника примерно у 90% эмбрионов.
Однако, когда матери подвергались умеренной гипоксии (8% кислорода) в течение 8 ч, только около 15% эмбрионов имели дефекты позвоночника, которые были очень легкими. Затем были объединены генетические и экологические модели, чтобы продемонстрировать, что эмбрионы мыши, гетерозиготные по нулевым мутантам генов сигнального пути Notch MESP2, HES7, DLL1 и Notch1 (исключая DLL3), показали повышенную восприимчивость и тяжесть дефектов позвоночника при комбинированном воздействии по сравнению с воздействием одного генетического фактора или фактора окружающей среды. Наконец, был исследован основной молекулярный механизм эмбриональной гипоксии, вызывающей дефекты позвоночника. Здесь было обнаружено, что гипоксия, по-видимому, прерывает передачу сигналов FGF в PSM. Это приводит к потере циклической активации передачи сигналов Notch, необходимой для образования сомитов, и, следовательно, к невозможности сегментации сомитов (рис. 2).
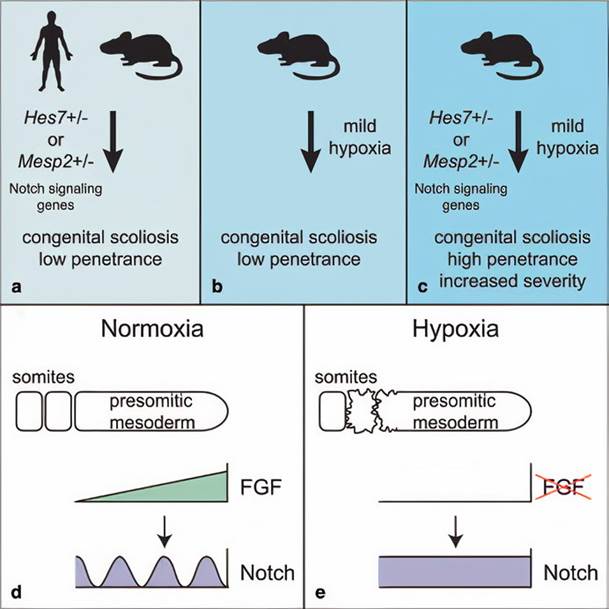
Рис. 2. a) Гетерозиготные мутации в генах Hes7 и Mesp2 пути Notch могут вызывать небольшие дефекты позвоночника у людей и мышей; б) Внутриутробное воздействие на эмбрионы мыши умеренными уровнями гипоксии может вызвать легкие дефекты позвоночника; c) Комбинация гетерозиготной мутации Hes7 или Mesp2 и внутриутробное воздействие на эмбрионы мыши умеренной гипоксии вызывают дефекты позвонков с повышенной пенетрантностью и степенью тяжести; d) В нормоксических условиях передача сигналов FGF присутствует в градиенте в PSM, и происходит циклическая активация передачи сигналов Notch. Правильная сегментация сомитов требует циклической активации передачи сигналов Notch; e) Гипоксия снижает общие уровни передачи сигналов FGF в PSM, и циклическая активация передачи сигналов Notch прекращается. Это приводит к аномальной сегментации сомитов [23]
Другие гены, влияющие на возникновение врожденных деформаций позвоночника
Помимо мутаций в генах, непосредственно участвующих в образовании сомитов, мутации в неспецифических генах, защищающих растущий эмбрион от воздействия тератогенных факторов, могут влиять на возникновение врожденных пороков развития и, следовательно, на возникновение ВДП. Такие генетические системы представляют собой барьерные системы детоксикации или биотрансформации ксенобиотиков, а также гены, которые непосредственно устраняют «сбои» в ДНК, – гены репарации ДНК. Такие гены, как CYP1A1, CYP1A2, GSTT1, GSTM1, NAT2, контролируют активность ферментов детоксикации, которые превращают мутагенные и тератогенные факторы окружающей среды в безвредные соединения. Мутации в этой группе генов могут препятствовать нейтрализации тератогенных факторов и приводить к мутагенезу de novo и, как следствие, к различным нарушениям эмбриогенеза, включая развитие врожденных аномалий позвоночника.
Важные гены репарации ДНК, такие как XRCC1 и XRCC3, также играют большую роль в устранении воздействия тератогенных факторов. Определение полиморфизма этих генов у пациентов с врожденными деформациями позвоночника может выявить первичные этиологические факторы данного патологического процесса и предопределить характер деформаций позвоночника в раннем возрасте.
Исследования генов детоксикации и репарации ранее проводились при различных наследственных и экзогенных заболеваниях. В частности, такие исследования чаще всего проводились у онкологических больных с опухолями различной этиологии, и была показана связь с предрасположенностью к заболеванию [41, 42]. Фактически изменение активности генов детоксикации и репарации приводит к тому, что в организме накапливается больше тератогенных факторов, которые могут токсически влиять на обмен веществ, вызывая повреждение органов и тканей. Это может также серьезно повлиять на организм беременной женщины и эмбриона. На эту тему есть публикации, в которых обсуждается взаимосвязь активности генов детоксикации и репарации с появлением врожденных пороков развития [43]. Мутации в генах репарации ДНК могут напрямую приводить к хромосомным и генным аномалиям. Поэтому изучение этиологии врожденных деформаций позвоночника неизбежно приводит к необходимости изучения генов детоксикации и репарации.
Наши результаты [44] указывают на корреляцию между мутационными изменениями в этих генах и возникновением врожденных деформаций позвоночника. В частности, значительные изменения в генах CYP1A2, GSTM1, GSTT1, NAT2, XRCC3 показали, что эти гены (а следовательно, и сама система детоксикации и репарации ДНК) участвуют в процессе защиты сомитогенеза от тератогенных нарушений. Если эта защитная система имеет бреши, то с большой долей вероятности могут возникнуть врожденные деформации позвоночника.
Это исследование подтверждает тот факт, что мутации в генах репарации ДНК могут приводить к хромосомным и генным аномалиям. Эти данные подтверждаются данными литературы [4, 45], в которых подчеркивается, что у большинства пациентов с врожденными деформациями позвоночника наблюдается сочетание пороков развития других внутренних органов и систем, что связано с хромосомными аберрациями в группе сцепления с другими генами данной группы.
Заключение
В последние годы исследователи добились значительного прогресса в понимании генетических факторов, лежащих в основе врожденных деформаций позвоночника, с наиболее важными мутациями, по крайней мере, в восьми генах, которые были идентифицированы на сегодняшний день. Этому прогрессу в значительной степени способствовали параллельные исследования на животных моделях, в частности на мышах. Такие модели важны для расчета генетических и сигнальных путей, необходимых для формирования паттерна тканей-предшественников позвонков (сомитов) в развивающемся эмбрионе. С помощью этих исследований был идентифицирован ряд генов-кандидатов для определения дефектов позвоночника у людей с использованием классических и новых генетических методов исследования. Наиболее важно то, что большинство генов, которые были обнаружены к настоящему времени и вызывают врожденные деформации позвоночника у людей, связаны с сигнальным путем Notch: DLL3 является ингибирующим лигандом; LFNG гликозилирует рецептор Notch; гены LFNG, MESP2, HES7 и TBX6 являются прямыми транскрипционными мишенями пути активации. Кроме того, белок HES7 подавляет передачу сигналов Notch, создавая петлю отрицательной обратной связи.
Появление полноэкзомного секвенирования с возможностью доступного полногеномного секвенирования в ближайшем будущем расширяет кругозор, но также создает новую проблему: будет происходить выявление многих потенциально повреждающих гетерозиготных генов и регуляторных вариантов у каждого человека, хотя только один или два из них могут вызвать деформацию позвоночника. Это потребует секвенирования нескольких больных и здоровых членов каждой семьи, что еще больше повысит важность функциональных исследований на животных моделях и системах in vitro для проверки патогенности вариантов. Недавние открытия, касающиеся влияния окружающей среды на формирование позвонков, добавляют еще один уровень сложности и в долгосрочной перспективе могут служить основой для терапевтического вмешательства, направленного на минимизацию тяжести и/или пенетрантности позвоночных дефектов в семьях с обнаруженными генетическими аномалиями. Тем не менее создание надежных генетических диагностических платформ для прогнозирования вариантов развития ВДП позволит назначать адекватное лечение этого тяжелого заболевания в раннем возрасте. Это дает реальную перспективу эффективных методов клинической генетики и надежду на излечение людей с врожденными пороками развития.
Библиографическая ссылка
Хальчицкий С.Е., Согоян М.В., Ли А.О., Мульдияров В.П., Кокушин Д.Н., Виссарионов С.В., Дмитриев А.В. ГЕНЕТИЧЕСКИЕ ФАКТОРЫ ЭТИОЛОГИИ И ПАТОГЕНЕЗА ВРОЖДЕННЫХ ДЕФОРМАЦИЙ ПОЗВОНОЧНИКА // Современные проблемы науки и образования. 2021. № 5. ;URL: https://science-education.ru/ru/article/view?id=31112 (дата обращения: 02.07.2026).
DOI: https://doi.org/10.17513/spno.31112