



CHED-синдром – крайне редкое наследственное двустороннее аутосомно-рецессивное заболевание, характеризующееся непрогрессирующим отеком роговицы с рождения. По данным литературы, встречаемость данного заболевания составляет 3:100 000 новорожденных, в сочетании с врожденной глаукомой – 6:100 000 новорожденных [1].
Ранее CHED-синдром разделялся на две формы: аутосомно-доминантную (CHED-1) и аутосомно-рецессивную (CHED-2). Однако последние данные генетического анализа и обзор клинических данных подтвердили, что ранее называвшаяся аутосомно-доминантной CHED (первоначально CHED-1) является формой задней полиморфной дистрофии роговицы с ранним и тяжелым отеком. На основании обновлений Международной классификации дистрофий роговицы 2015 г. термин «CHED» теперь относится исключительно к аутосомно-рецессивному CHED (первоначально CHED-2) [1–3].
В литературе имеются сообщения взаимосвязи данного заболевания с миссенс-мутацией гена slc4a11 на хромосоме 20, локусе р15, относящегося к группе генов SLC4 (Solute carrier 4), кодирующего 9 мембранных белков, преимущественно участвующих в транспорте бикарбоната (бикарбонатная буферная система). Белки SLC4 играют важную роль во многих режимах кислотно-основного гомеостаза [4, 5].
Мутации данного гена выявляются при врожденной наследственной эндотелиальной дистрофии, синдроме Харбояна и при ранней форме эндотелиальной дистрофии роговицы Фукса. Молекулярное действие и роль данного гена в здоровой и больной роговице в настоящее время недостаточно изучены [6–8].
Первоначально сообщалось, что SLC4A11 (первоначально названный BTR1) транспортирует борат, связанный с Na+. Однако сегодня SLC4A11 считается многофункциональным транспортером, который опосредует поток H+, NH3 [9–11]. Есть предположения, что продукты гена участвуют в регулировании уровней азота в тканях [4, 7, 12].
В зарубежной литературе имеются сообщения о сопутствующей врожденной глаукоме у пациентов с CHED-синдромом. Следует уточнить, что эндотелий роговицы и трабекулярная сеть имеют общее эмбриональное происхождение из нервного гребня концевых отделов нервного желобка [5, 13]. Однако, по данным других авторов, экспрессия SLC4A11 не была продемонстрирована в структурах, формирующих отток внутриглазной жидкости [3, 14].
Клинические примеры. В настоящем сообщении представлены клинические случаи двух родных братьев с врожденной наследственной эндотелиальной дистрофией роговицы, наблюдавшихся в Калужском филиале ФГАУ «НМИЦ «МНТК “Микрохирургия глаза” имени академика С.Н. Федорова» Минздрава России.
Пациент С., 2004 года рождения (старший брат), наблюдается в клинике с возраста 5 месяцев. Причина обращения – помутнение роговиц обоих глаз сразу после рождения. При первичном обращении отмечались выраженный отек роговиц, утолщение оптического среза роговицы (OD > OS), незначительное увеличение диаметра роговиц, передняя камера средней глубины, влага прозрачная, радужная оболочка структурная, хрусталик прозрачный. По данным пахиметрии толщина роговицы достигала 1006 мкм на правом глазу и 1010 мкм на левом, также выявлялось повышенное внутриглазное давление – 25 мм рт. ст. на правом и 27 мм рт. ст. на левом глазу, увеличенный диаметр роговицы: горизонтальный размер – 11,0 мм, вертикальный размер – 11,5 мм, что превышает возрастную норму. ПЗО: OD – 21,02 мм, OS – 20,98 мм. Кератометрия: OD AX: 5 -> 33,37; AX: 95 -> 37,00; OS AX: 180 -> 41,87; AX: 90 -> 45,62. При проведении УЗ-биомикроскопии определялась мезенхимальная ткань в углу передней камеры – гониодисгенез 1–2-й степени, глубина передней камеры составляла: OD – 3,34 мм, OS – 3,24 мм.
Пациенту был поставлен диагноз «врожденная глаукома, ассоциированная с дистрофией роговицы». В возрасте 7 месяцев была проведена антиглаукоматозная операция в связи с некомпенсированным внутриглазным давлением и клиническими проявлениями врожденной глаукомы. В послеоперационном периоде внутриглазное давление находилось в пределах нормы, данные ПЗО и диаметр роговицы увеличивались в пределах физиологических значений, однако отек и толщина роговиц обоих глаз оставались прежними. На основании двухстороннего симметричного характера процесса предположили наследственное заболевание роговицы, в связи с чем пациент был направлен на генетическое обследование.
Пациент наблюдался в Калужском филиале МНТК до 2-летнего возраста. При обследовании внутриглазное давление колебалось в пределах от 18 до 20 мм рт. ст., сохранялась прежняя толщина роговицы, которая по данным пахиметрии составляла 1050 мкм, сохранялся ее стромальный отек.
В дальнейшем, вплоть до 2014 г., пациент С. не посещал клинику, и очередной визит состоялся уже вместе с маленьким братом, рожденным в 2014 г., у которого выявлялась идентичная клиническая картина. Генетический анализ на тот момент так и не был проведен.
При обследовании в 2014 г. у пациента С. (старшего брата) по-прежнему определялся отек роговиц, более выраженный на правом глазу. Острота зрения с коррекцией правого глаза составляла 0,2, левого глаза – 0,8. По данным ОСТ определялись увеличенная толщина роговицы правого глаза до 1150 мкм, усиление рефлективности и утолщение на уровне боуменовой мембраны. По данным электронной микроскопии отмечались полиморфизм, полимегетизм клеток, преобладающий размер клеток составлял 400 нм. Было принято решение о проведении патогенетически обоснованного хирургического лечения.
В 2016 г. проведена задняя фемтолазерная послойная кератопластика с ультратонким трансплантатом на правом глазу. Через 1 месяц после операции корригированная острота зрения правого глаза повысилась до 0,4, толщина роговицы по данным ОСТ уменьшилась до 824 мкм (рис. 1а, 1б), при исследовании плотности эндотелиальных клеток их количество составило 2350 кл/мм². По данным компьютерной периметрии поле зрения правого глаза было в норме, обследование левого глаза затруднено. Субъективно пациент отмечал повышение четкости изображения и увеличение контрастности цветов.
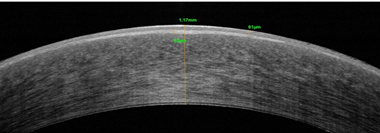
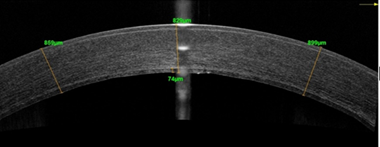
Рис. 1. Пациент С. ОСТ роговицы до (а) и через 1 месяц после (б) задней фемтолазерной послойной кератопластики с ультратонким трансплантатом на правом глазу
Пациент Е., 2014 года рождения, родной младший брат пациента С., поступил в клинику в возрасте 3 месяцев с идентичными жалобами матери на помутнение роговиц с рождения.
Биомикроскопически определялось незначительное увеличение диаметра роговицы, выявлялись стромальный отек, увеличенный оптический срез роговицы. Так же, как у брата, выявлялись клинические признаки врожденной глаукомы – повышенное внутриглазное давление до 25 мм рт. ст., увеличенный диаметр роговицы: горизонтальный размер – 11,5 мм, вертикальный размер – 11,5 мм, что превышает возрастную норму. ПЗО: OD – 20,95 мм, OS – 21,2 мм. При проведении УЗ-биомикроскопии определялась мезенхимальная ткань в углу передней камеры – гониодисгенез 1–2-й степени, глубина передней камеры составляла: OD – 3,34 мм, OS – 3,24 мм. Кератометрия: OD AX: 5 -> 43,5; AX: 95 -> 47,00 OS AX: 161 -> 43,25; AX: 71 -> 47,0. По данным ОСТ толщина роговицы обоих глаз составляла 1050 мкм. Пациенту был поставлен диагноз «эндотелиальная дистрофия роговицы, ассоциированная врожденная глаукома». В возрасте 6 месяцев проведена антиглаукоматозная операция на левом глазу в связи с отсутствием компенсации ВГД на фоне медикаментозной терапии.
В очередной раз оба брата появились в клинике уже в 2019 г. Выяснилось, что пациенты обращались в немецкую клинику, где был проведен молекулярно-генетический анализ гена SLC4A11, по результатам которого выявлены гетерозиготные мутации в экзоне 10 с.1237G>A, р.(Gly413Arg) и с.1361Т>С, р.(Leu454Pro) в экзоне 11-го гена SLC4A11. Полученные данные позволили утвердиться в диагнозе «CHED-синдром».
В 2020 г. у пациента С. появились жалобы на ухудшение зрения левого глаза. При обследовании биомикроскопически отмечалось увеличение отека роговицы левого глаза; корригированная острота зрения правого глаза составляла 0,5, левого глаза снизилась на 2 строчки и составила 0,6. По данным ОСТ роговицы толщина роговицы правого глаза составляла 824 мкм (толщина трансплантанта 76 мкм), левого глаза – 1140 мкм.
Было принято решение о трансплантации десцеметовой мембраны со слоем эндотелиальных клеток. Через 1 месяц после операции острота зрения левого глаза повысилась на 1 строчку и составила 0,3 с коррекцией, субъективно пациент отмечал увеличение контрастности и яркости цветов. По данным ОСТ роговицы наблюдались значительное снижение ее толщины (до 643 мкм (рис. 2а, 2б)), фиброплазия боуменовой мембраны и усиление рефлективности передних слоев стромы, также обращала на себя внимание выраженная иррегулярность боуменовой мембраны в виде ее складчатости, однако степень иррегулярности постепенно уменьшалась к сроку наблюдения 3 и 6 месяцев. Такую иррегулярность мы связываем с резкой резорбцией отека и невозможностью боуменовой мембраны к сокращению.
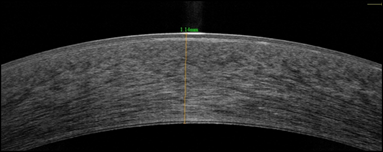
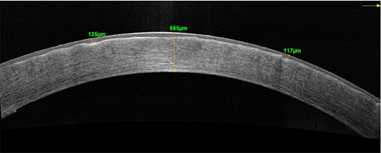
Рис. 2. Пациент С. ОСТ роговицы до (а) и через 1 месяц после (б) трансплантации десцеметовой мембраны со слоем эндотелиальных клеток
У пациента Е. (младшего брата) в 2020 г. в возрасте 6 лет отмечалось ухудшение зрения правого глаза, что сопровождалось увеличением степени отека роговицы. Было принято решение о проведении трансплантации десцеметовой мембраны. После операции толщина роговицы уменьшилась до 816 мкм, роговица стала оптически прозрачна. В послеоперационном периоде через 1 (рис. 3а, 3б) и через 3 месяца сохранялись утолщение и усиление рефлективности на уровне боуменовой мембраны, однако ее иррегулярности, как у старшего брата, не выявлялось.
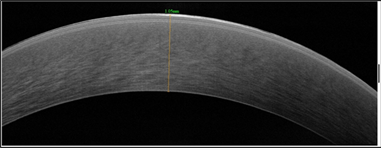
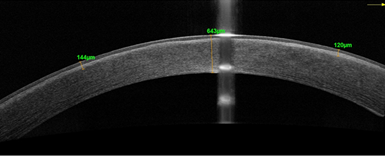
Рис. 3. Пациент Е. ОСТ роговицы до (а) и через 1 месяц после (б) трансплантации десцеметовой мембраны со слоем эндотелиальных клеток
В итоге два брата с диагнозом «CHED-синдром» были прооперированы в различные возрастные периоды и с применением различных технологий.
У младшего брата трансплантация десцеметовой мембраны в возрасте 6 лет привела к значительному уменьшению толщины роговицы. Аналогичная операция у старшего брата в возрасте 16 лет также привела к значительному снижению толщины роговицы, однако менее выраженному в сравнении с динамикой резорбции отека у младшего брата, а также выявлены изменения передней поверхности роговицы в виде иррегулярности боуменовой мембраны.
У старшего брата задняя послойная кератопластика с ультратонким трансплантатом в возрасте 12 лет привела к менее выраженным изменениям толщины роговицы, но иррегулярности роговицы не отмечалось.
Заключение. Таким образом, на основании анализа двух клинических случаев ведения пациентов с CHED-синдромом можно предположить, что:
- трансплантация десцеметовой мембраны со слоем эндотелиальных клеток при CHED-синдроме характеризуется большей резорбцией отека стромы роговицы, чем при UTFS-dsek;
- трансплантация десцеметовой мембраны, выполняемая на ранних сроках, характеризуется восстановлением физиологической толщины роговицы;
- трансплантация десцеметовой мембраны, проведенная на поздних сроках, может приводить к развитию иррегулярности на уровне боуменовой мембраны в ранние сроки послеоперационного периода.
Требуется дальнейшее подтверждение полученных результатов на большем клиническом материале.
Библиографическая ссылка
Терещенко А.В., Демьянченко С.К., Трифаненкова Я.М. ЭНДОТЕЛИАЛЬНАЯ КЕРАТОПЛАСТИКА ПРИ ВРОЖДЕННОЙ НАСЛЕДСТВЕННОЙ ЭНДОТЕЛИАЛЬНОЙ ДИСТРОФИИ: 2 СЛУЧАЯ В ОДНОЙ СЕМЬЕ // Современные проблемы науки и образования. 2021. № 3. ;URL: https://science-education.ru/ru/article/view?id=30694 (дата обращения: 27.06.2026).
DOI: https://doi.org/10.17513/spno.30694