



Синдром Жёна, или торакоасфиктическая дисплазия грудной клетки, – чрезвычайно редкая наследственная патология с аутосомно-рецессивным типом наследования, диагностируемая с частотой 1:100 000 – 1:130 000 новорожденных. Заболевание характеризуется узкой колоколообразной грудной клеткой с короткими толстыми горизонтально расположенными ребрами, что приводит к возникновению гипоплазии легких и легочной недостаточности после рождения. В 1955 г. французский педиатр M. Jeune описал случай семейной удушающей грудной дистрофии у пары братьев и сестер с сильно узкой грудной клеткой, и данная патология в последующем стала известна как синдром Jeune (Жёна, Жене) [1]. Синдром Жёна – потенциально смертельная врожденная карликовость, которая характеризуется наличием типичных скелетных дисплазий (узкая деформированная грудная клетка, микромелия), а также поражениями внутренних органов (почек, печени, ЦНС). Респираторные нарушения широко варьируют от тяжелой дыхательной недостаточности и смерти до инфантильного латентного фенотипа без респираторных проявлений [2]. Синдром Жёна можно обнаружить как антенатально, так и при рождении или в раннем детстве из-за типичных ультразвуковых, клинических и рентгенологических признаков.
Синдром Жёна – генетически гетерогенное расстройство, являющееся представителем цилиопатий, или расстройств, связанных с дисфункциями первичных ресничек [3, 4, 5]. В 2013 г. Шмидт обнаружил 34 мутации в гене DYNC2H1 у 41% пациентов с данной патологией, что позволило считать наличие мутаций в этом гене основной причиной развития синдрома Жёна. В настоящее время известны и другие гены, мутации которых могут приводить к рождению ребенка с проявлениями синдрома Жёна: IFT80, TTC21B/IFT139, IFT140, WDR19/IFT1448, расположенные в локусах хромосом 3q25.33, 11q25.3, 4p14, 2q24.3, 15q13. Все они кодируют белки, принадлежащие семейству IFT (Intraflagellar transport), имеющие большое значение для цилиогенеза, которые управляют целым рядом важных клеточных событий в нормальном развитии человека. Учитывая разнообразие вовлеченных в мутации генов и различные формы мутаций (сдвиг рамки считывания, делеции, сайт-сплайсинг), количество вариаций мутантных генов может достигать более семидесяти [4].
Описание клинического случая
Ребенок К. (мальчик) от матери, жительницы Омской области, с неотягощенной наследственностью, без вредных привычек. Данная беременность третья (предыдущие две закончились рождением доношенных здоровых мальчиков), протекала на фоне эрозии шейки матки. На учете в женской консультации с 16 недель, первая половина беременности, со слов матери, протекала относительно благоприятно. В 18 недель беременности по УЗИ были выявлены признаки аномалии развития скелета у плода: укорочение трубчатых костей, искривление и деформация костей предплечья, снижение оссификации костей свода черепа, гипоплазия костей носа, долихоцефалическая форма головки. Пренатальным консилиумом дальнейшее вынашивание беременности не рекомендовано ввиду неблагоприятного прогноза с высоким риском летального исхода, однако семья категорически отказалась от прерывания беременности.
Ребенок родился в Омском областном перинатальном центре в 39 недель гестации путем планового кесарева сечения, с оценкой по шкале Апгар 7/7 баллов, в реанимационных мероприятиях при рождении не нуждался. Однако уже в первые часы у ребенка начала нарастать дыхательная недостаточность, по поводу чего новорожденный в возрасте трех часов жизни переведен на искусственную вентиляцию легких (ИВЛ).
При осмотре ребенка (рис. 1) обращали внимание диспропорциональное телосложение (укорочение конечностей), наличие множественных деформаций грудной клетки, мягкие кости черепа с открытыми швами. В первые часы жизни манифестировал геморрагический синдром (легочное и желудочно-кишечное кровотечение).
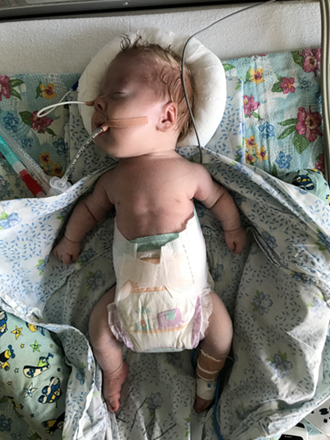
Рис. 1. Внешний вид новорожденного с фенотипом синдрома Жёна
В клинической картине преобладали симптомы дыхательной недостаточности, а также поражения центральной нервной системы (симптомы угнетения, мышечная дистония, псевдобульбарный синдром).
Рис. 2. Рентгенограмма грудной клетки новорожденного К.
Дыхание с первых суток жизни с аппаратной поддержкой, проводилась искусственная вентиляция легких. При аускультации в легких выслушивались крепитация, влажные хрипы, при санации дыхательных путей отделялось большое количество жидкой светлой мокроты (возможное проявление цилиарной недостаточности в виде нарушения мукоцилиарного клиренса). Самостоятельное дыхание неэффективное, попытки спонтанного дыхания нерегулярные.
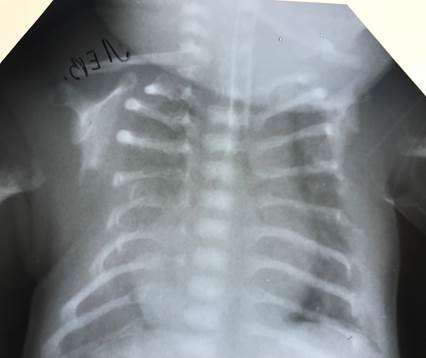
На рентгенограмме органов грудной клетки в первые сутки определялись интерстициальные изменения в легочной ткани, характерные для респираторного дистресс-синдрома (РДС), которые в последующем сменились ателектазами легочной ткани; помимо этого, на рентгенограмме отчетливо определялись костные изменения (рис. 2).
В газовом составе крови определялась стойкая гиперкапния с респираторным ацидозом, что свидетельствовало о неэффективности внешнего дыхания, обусловленной скелетными деформациями грудной клетки.
При выполнении мультиспиральной компьютерной томографии (МСКТ) грудной клетки описывались изменения в легких, которые расценивались как проявление РДС на фоне неполного расправления легочной ткани; субплевральные ателектазы в S3 правого легкого и S6, S10 левого легкого, а также костные нарушения (рис. 3): задержка костного возраста, порок развития длинных трубчатых костей верхних конечностей (гипоплазия метаэпифизов обеих плечевых костей, костей предплечий, гипоплазия суставных поверхностей обеих лопаток – аплазия поперечная терминальная).
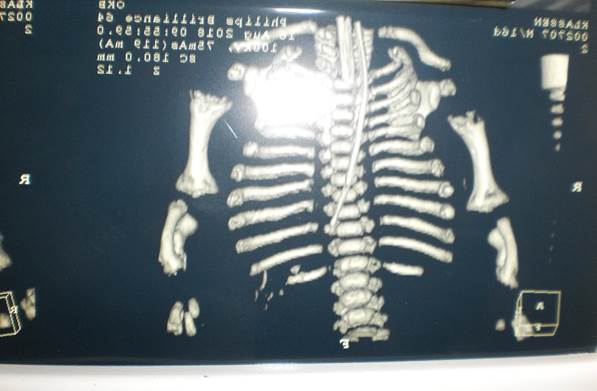
Рис. 3. МСКТ грудной клетки новорожденного К.
При исследовании сердца с помощью эхокардиографии были выявлены умеренно выраженные признаки легочной гипертензии, что также укладывается в симптоматику данного синдрома.
При ультразвуковом исследовании внутренних органов, помимо спленомегалии, других изменений выявлено не было, хотя синдром Жёна может иметь в своих проявлениях поражение внутренних органов (печени, почек). Однако из данных литературы известно, что изменения во внутренних органах с развитием клиники почечной и печеночной недостаточности происходят уже в постнеонатальном периоде (1–2-й год жизни) [6, 7, 8].
По нейросонографии у ребенка были описаны ультразвуковые признаки незначительной вентрикулодилятации, отека перивентрикулярных тканей и незрелости ЦНС, при этом отек перивентрикулярных тканей сохранялся длительно.
В общем анализе крови на первой неделе жизни отмечались воспалительные изменения, было повышение С-реактивного белка до 72 г/л, что наряду с клиническими проявлениями (такими как дыхательная недостаточность, влажные хрипы в легких, обилие мокроты при санации дыхательных путей) позволило выставить диагноз пневмонии, при этом рентгенологические изменения в легких были слабо выражены.
Учитывая внешние проявления, ребенку было проведено генетическое исследование (определение кариотипа и скрининг на основные мутации, при которых возможны данные фенотипические проявления). Хромосомные аберрации не были выявлены, кариотип ребенка соответствовал 46ХУ.
В условиях цитогенетической лаборатории медико-генетической консультации БУЗОО «Областная клиническая больница» г. Омска было проведено молекулярно-цитогенетическое исследование крови (гибридизация зондов с ДНК интерфазных ядер) на поиск мутаций в причинно-значимых генах: мутация в гене 1q21/SRD (делеции не выявлено), Cri-Du-Chat CTNND2(5P15)/5Q31 (делеции не выявлено), синдром Сотоса NSD1(5q35)/TERT(5p15) (делеции не выявлено), Wolf–Hirschhorn WHSC1(4p16)/SE4 (делеции не выявлено), Williams–Beuren ELN(7q11)/7q22 (делеции не выявлено), синдром Ди-Джорджи (делеции не выявлено), синдром Ангельмана (делеции не выявлено), синдром Прадера–Вилли (делеции не выявлено), синдром Миллера–Дикера (делеции не выявлено). Несмотря на наличие ярких клинических проявлений, соответствующих фенотипу синдрома Жёна, проведенное молекулярно-цитогенетическое исследование не выявило наиболее распространенных мутаций, характерных для данной патологии. Учитывая, что количество возможных мутаций при данном синдроме может достигать 77, необходимо было проведение расширенного исследования с использованием генетических панелей. К сожалению, в условиях медико-генетической консультации Омской областной клинической больницы данное исследование было недоступно.
В возрасте 27 дней жизни, учитывая неблагоприятный прогноз, ребенок был переведен в отделение паллиативной помощи центральной районной больницы Кормиловского района Омской области, где дыхательная недостаточность прогрессивно нарастала. В возрасте 1 месяца ребенок умер.
Основной диагноз: Системная патология скелета в виде асфиктической дистрофии Жёна. Перинатальное постгипоксически-ишемическое поражение ЦНС, незначительная вентрикулодилятация, синдром угнетения центральной нервной системы с переходом в синдром возбуждения, псевдобульбарный синдром. Угроза задержки психомоторного развития.
Осложнение основного: Неполное расправление легочной ткани, субплевральные ателектазы S3 справа, S6 и 10 слева. Анемия постгеморрагическая тяжелой степени (гемотрансфузия № 1). Ретинальные кровоизлияния обоих глаз.
Сопутствующий диагноз: Транзиторное тахипноэ новорожденного в анамнезе. Внутриутробная инфекция неуточненной этиологии (пневмония) на фоне врожденного порока развития скелета и легких. Геморрагический синдром (легочное, желудочное кровотечение) в анамнезе. Гипокоагуляция новорожденного в анамнезе (плазмотрансфузия № 2). Персистирующая легочная гипертензия в анамнезе.
Фоновое: Морфофункциональная незрелость.
Обсуждение. Синдром Жёна – редкое заболевание, часто не известное широкому кругу врачей. В настоящее время в отечественной педиатрии имеются сообщения о серии наблюдений за детьми, которым диагностирована данная патология (7 человек без учета нашего пациента) [1, 9]. В наблюдениях Д.Ю. Овсянникова (2016) характерным проявлением данного синдрома было сочетание таких симптомов, как отставание в росте грудной клетки, укорочение конечностей, дыхательная недостаточность с кислородозависимостью, рецидивирующие инфекции дыхательных путей, задержка моторного развития. Поскольку в основе заболевания лежит генетически обусловленное нарушение структуры или функции ресничек (цилий), которые принимают участие в развитии костной, гепатобилиарной систем, нефронов, сетчатки глаза и других структур организма, то и фенотипические проявления данного синдрома могут быть самыми разнообразными [10]. Цилиопатии являются плейотропными заболеваниями и характеризуются такими общими, вариабельными в зависимости от заболевания клиническими симптомами, как поликистоз почек, печени и поджелудочной железы, дегенерация сетчатки глаза, асимметрия внутренних органов, костные, челюстно-лицевые дефекты и дефекты одонтогенеза, дефекты нервной системы, гидроцефалия, ожирение, бесплодие. В эмбриональном периоде цилии необходимы для правильного межклеточного взаимодействия в процессе остеохондрогенеза, поэтому наиболее частым и ранним проявлением синдрома Жёна является системная остеохондродисплазия, которую можно выявить уже антенатально.
Отдельного внимания заслуживает патогенез развития дыхательной недостаточности при синдроме Жёна. Как показывает демонстрируемый клинический случай, достаточно тяжелый респираторный дистресс-синдром способен развиться даже у доношенного зрелого ребенка. Патогенез дыхательных нарушений при торакоасфиктической дисплазии во многом связан с деформацией грудной клетки, которая происходит внутриутробно и нарушает дыхательную активность плода, которая в свою очередь является важным фактором нормального развития легких. Дыхательные движения плода представляют собой периодические ритмичные сокращения диафрагмы, которые поддерживают растяжение легких и объем жидкости в периоды отдыха при сокращении верхних дыхательных путей. Отсутствие дыхательных движений у плода способно приводить к гипоплазии легких. При синдроме Жёна движение грудной клетки затруднено вследствие костных деформаций, что приводит к уменьшению дыхательных движений, а это в свою очередь способствует формированию гипоплазии легких различной степени выраженности [11, 12]. После рождения прогрессирование дыхательной недостаточности происходит в результате совокупности причин: нарушение внешнего дыхания из-за скелетных деформаций и нарушение диффузии газов в легочной паренхиме вследствие ее гипоплазии. Помимо этого, уже в раннем периоде может манифестировать цилиарная недостаточность в виде нарушения мукоцилиарного клиренса и развития пневмонии. Дыхательная недостаточность способна значительно усугубляться сердечно-сосудистой патологией – легочной гипертензией (является частым спутником гипоплазированных легких), врожденными пороками сердца с обогащением малого круга кровообращения [9]. Таким образом, скелетные аномалии и поражение респираторной системы служат основными клиническими проявлениями синдрома Жёна. Одним из паллиативных способов лечения при синдроме Жёна является хирургическое устранение деформации грудной клетки, что в ряде случаев позволяет добиться некоторого улучшения роста легких. Однако данная методика малоэффективна при выраженной гипоплазии легочной паренхимы, поскольку шансы на улучшение прогноза в данной ситуации невелики.
Вместе с тем при данном заболевании может развиваться различной степени выраженности патология других органов. При синдроме Жёна поражаются почки, печень, поджелудочная железа, реже – желудочно-кишечный тракт, нервная система и глаза [6, 7].
Поражение почек чаще развивается после первых 2 лет жизни вследствие неправильного восприятия сигналов, необходимых для роста и развития почечной ткани из-за нарушенной работы цилий в клетках эпителия нефрона. Это приводит к тубулоинтерстициальной нефропатии, атрофии и кистозным изменениям в паренхиме почек, нарушению функции канальцевого аппарата, диффузному интерстициальному фиброзу, гломерулярному склерозу [8]. Почечная недостаточность – основная причина летального исхода у пациентов с синдромом Жёна в возрасте от 3 до 10 лет [12]. Патология печени возникает в результате неправильного развития протоковой пластинки в период эмбриогенеза. Протоковая пластинка образуется из клеток печени, расположенных вокруг сосудов, и обеспечивает формирование нормальной архитектоники печени. Некоторые структурные изменения печени, такие как поликистоз, пролиферация эпителия желчных протоков и портальный фиброз, как правило, протекают бессимптомно и обнаруживаются только при ультразвуковом исследовании (УЗИ) или на аутопсии, однако при синдроме Жёна могут возникать и клинически значимые нарушения, включающие гепатомегалию, портальную гипертензию, цирроз, холестаз [6].
Заключение. Таким образом, наличие фенотипических проявлений у новорожденного К. укладывается в клиническую картину синдрома Жёна, который имеет в большинстве случаев неблагоприятный прогноз. Известно, что летальность в первые 2 года жизни у больных с данной патологией составляет 60−80%, при этом большинство летальных исходов регистрируется в неонатальном периоде [1, 9]. У представленного нами пациента преобладали респираторные проявления синдрома Жёна (выраженная деформация грудной клетки, обусловившая глубокие нарушения вентиляционной функции), что и явилось причиной неблагоприятного исхода. При менее выраженной деформации скелета ребенок с данной патологией может пережить младенческий возраст, и тогда в клинической картине будет преобладать симптоматика поражения внутренних органов, прежде всего почек. В целом выраженность клинических проявлений синдрома Жёна определяется типом мутации генов, вовлечением поражения скелета (прежде всего грудной клетки), а в последующие возрастные периоды – и других органов и систем.
Библиографическая ссылка
Белкова Т.Н., Оксеньчук Т.В., Кривцова Л.А., Каташова Е.Н., Афанасьев М.В., Храпов Д.В. КЛИНИЧЕСКИЙ СЛУЧАЙ СИНДРОМА ЖЁНА У НОВОРОЖДЕННОГО // Современные проблемы науки и образования. 2020. № 2. ;URL: https://science-education.ru/ru/article/view?id=29753 (дата обращения: 04.07.2026).
DOI: https://doi.org/10.17513/spno.29753