



Расстройство аутического спектра (РАС) является одной из актуальных проблем современной психиатрии и неврологии ввиду высокой частоты встречаемости, клинической и генетической гетерогенности (таблица 1). РАС – расстройство нейропсихического развития, обычно наблюдаемое у детей, характеризующееся специфическим поведенческим фенотипом, сниженной социализацией и стереотипными поведенческими реакциями [1; 2]. По данным Всемирной организации здравоохранения (ВОЗ), оценки распространенности РАС известны по Европейскому региону и региону стран Америки, которые не различаются между собой в статистическом отношении: для Европы медианный показатель составляет 61,9:10000 (диапазон 30,0 – 116,1/10000), а для стран Америки – 65,5:10000 (диапазон 34,0 – 90,0/10000). Соотношение встречаемости РАС у мальчиков и у девочек находится в пределах от 2,6:1 до 4:1 [3].
По мере бурного развития медицинской генетики в последние годы показано, что РАС могут быть ассоциированы с широким кругом хромосомных [4; 5], моногенных [6-8], мультифакторных [9; 10] заболеваний.
Поэтому дифференциальная диагностика РАС в повседневной клинической практике является сложной задачей и требует от врача невролога и психиатра не только хороших навыков фенотипирования, но и включения в арсенал диагностических мероприятий современных молекулярно-генетических и цитологических методов диагностики. На фоне бурного развития медицинской генетики и трансляции её достижений в клиническую практику расширилось наше представление о РАС, а в структуру заболеваний, имеющих фенотип РАС, включаются всё новые и новые нозологии. К одному из недавно описанных заболеваний группы РАС относится синдром Люскан-Люмиш (СЛЛ), описанный Армеллией Люскан (2014) [7] и Хейди С. Люмиш (2015) [8].
Целью данного обзора является освещение редкого фенотипа РАС – синдрома Люскан-Люмиш для повышения настороженности практикующих врачей неврологов и психиатров в ранней диагностике СЛЛ у детей дошкольного возраста; также в статье рассматривается оценка важности трансляции современных методов молекулярно-генетической диагностики при ведении детей с РАС на примере СЛЛ.
Дефиниция
Синдром Люскан-Люмиш (OMIM: 616831) – синдром чрезмерного роста, развивающийся в результате мутации гена SETD2, и характеризуется макроцефалией, задержкой интеллектуального и речевого развития, низким уровнем социализации, расстройством аутистического спектра (РАС). Более вариабельные характеристики включают: постнатальный чрезмерный рост, ожирение, избыточную карпальную оссификацию, задержку психомоторного развития, эпилептические припадки [7; 8]. Кроме того, выделяют СЛЛ с Сотос-подобным фенотипом, для которого характерны вышеописанные клинические проявления, но без чрезмерного роста и избыточного веса [11].
Краткая история
Впервые мутация гена SETD2 на хромосоме 3p21 была описана B.J. O'Roak и соавт. (2012) при экзомном секвенировании образцов дезоксирибонуклеиновой кислоты (ДНК) членов семей со спорадическими случаями РАС (всего выявлено 2 мутации среди 677 больных РАС) [12; 13]. Iossifov I. и соавт. (2014) при экзомном секвенировании более 2500 семей, в которых были дети с РАС, также выявили двух пациентов с гетерозиготной мутацией (миссенс, делеция) в гене SETD2 [14].
Luscan A. и соавт. (2014), используя таргетное секвенирование нового поколения, при исследовании ДНК пациентов с синдромом Вивера (OMIM: 277590), Сотоса (OMIM: 117550), и Сотос-подобного синдрома (OMIM: 612778.0002, 612778.0003) выявили мутацию (нонсенс, миссенс) в гене SETD2 [7]. Годом спустя H.S. Lumish и соавт. (2015) при полноэкзомном секвенировании ДНК 17-летней девушки с синдромом Люскан-Люмиш выявили фреймшифт мутацию de novo в гене SETD2 [8], а позднее van M.C. Rij и соавт. (2018) описали два новых случая заболевания [11].
Патогенез
SETD2 – это фермент гистоновая метилтрансфераза, который триметилирует лизин гистона H3K36me3 в нуклеосомах (рисунок 1), являющегося специфическим ярлыком для эпигенетической активации транскрипции [15]. Считается, что данный фермент помогает восстановить нормальную структуру хроматина после транскрипции, тем самым подавляя ложную внутригенную транскрипцию. Он является ключевым регулятором восстановления несоответствия цепей дезоксирибонуклеиновой кислоты (ДНК) с помощью гомологичной рекомбинации в G1 и ранней S-фазе путем генерации H3K36me3; необходим для восстановления двухцепочечной поломки ДНК в ответ на её повреждение. Кроме того, он участвует в регуляции альтернативного сплайсинга [8; 16].
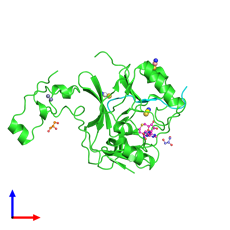
Рис. 1. Кристаллическая структура белка SETD2, связанного с пептидом гистона H3K3 [17]. Примечания: депонированная структура SETD2, связанного с пептидом гистона H3K3, раскрашенная по цепочкам. Вид спереди: 1) одна копия гистон-лизин-N-метилтрансферазы SETD2; 2) одна копия гистона H3.3; 3) четыре неполимерных объекта (1 копия S-аденозил-L-гомоцистеина, 3 копии иона тиоционата, 2 копии глицерола, 3 копии иона цинка)
Помимо вышеуказанных ключевых функций, SETD2 участвует в других биологических процессах: стимулирует дифференцировку эмбриональных стволовых клеток в направлении энтодермы; вместе с гистонами управляет метилированием других белков, таких как тубулины и STAT1 [18]; вовлечен в интерферон-альфа-индуцированную антивирусную защиту, катализируя H3K36me3 на промоторах некоторых интерферон-стимулированных генов для активации транскрипции генов [19].
Биологические процессы, в которые вовлечен фермент SETD2: регуляция восстановления двухцепочечного разрыва ДНК с помощью гомологичной рекомбинации [20]; регулирование экспорта матричной рибонуклеиновой кислоты (мРНК) из ядра; регуляция локализации белка в хроматине; регуляция транскрипции; нуклеосомная организация; организация цитоскелета микротрубочек, участвующих в митозе; дифференцировка энтодермальных клеток; морфогенез мезодермы; развитие стволовых клеток; дифференцировка стволовых клеток; эмбриональный морфогенез плаценты; эмбриональный морфогенез черепного скелета; развитие переднего мозга; закрытие нервной трубки; морфогенез коронарной сосудистой сети; развитие перикарда; положительная регуляция продукции интерферона-альфа; регуляция цитокинеза [16].
Фермент SETD2 кодируется геном SETD2 (альтернативные названия: HYPB; HBP231; HSPC069; HIF-1; HIP-1; KIAA1732; FLJ23184; KMT3A) [21], который располагается на хромосоме 3p21.31 (рисунок 2), и включает 26 экзонов.
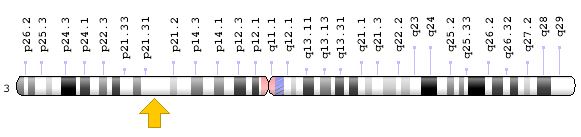
Рис. 2. Локализация гена SETD2 на хромосоме 3p21.31
Описано несколько заболеваний, которые могут быть вызваны различными мутациями, затрагивающими ген SETD2:
1) синдром Люскан-Люмиш (OMIM:616831); заболевание возникает при гетерозиготной мутации в гене SETD2; тип наследования заболевания – аутосомно-доминантный [8];
2) почечно-клеточная карцинома (OMIM:144700); дефекты SETD2 связаны с прекращением метилирования ДНК в непромоторных областях, что приводит к аберрантному и уменьшенному уплотнению нуклеосом и хроматиновой ассоциации ключевых белков репликации, таких как MCM7 и дельта ДНК-полимеразы, что в свою очередь замедляет развитие вилки репликации, делая невозможной гомологичную рекомбинацию при разрывах ДНК [22];
3) острый лимфобластный лейкоз (OMIM:613065); заболевание может быть вызвано мутациями, затрагивающими различные генетические локусы, включая ген SETD2 [23].
В настоящее время описаны следующие типы мутаций: нонсенс (образование неинформативных последовательностей и невозможность процесса транскрипции или синтез укороченных полипептидных цепей с нарушенной функцией); миссенс (изменение последовательности нуклеотидов в кодоне, что приводит к изменению состава аминокислот в синтезируемом полипептиде); фреймшифт (мутация сдвига рамки считывания - тип мутации в последовательности ДНК, для которого характерна вставка или делеция нуклеотидов, в количестве не кратном трём; в результате происходит сдвиг рамки считывания при транскрипции матричной рибонуклеиновой кислоты (мРНК)) [11-13]; делеция (хромосомная аберрация, при которой выпадает внутренний участок хромосомы) [14].
Выделяют 4 аллельных варианта СЛЛ (таблица 1), которые внесены в базу данных Online Mendelian Inheritance in Man (OMIM) [24], однако по мере изучения генетики заболевания число патогенных мутаций, ответственных за фенотип СЛЛ и зарегистрированных в базе данных MalaCards: The human disease database [25], превышает 250, что может затруднять поиск патогенных мутаций у конкретного ребёнка с фенотипом РАС. Однако внедрение современных методов секвенирования позволяет провести поиск не только ранее описанных мутаций, но и новых, выявляемых в раннем диагностическом поиске.
Таблица 1
Аллельные варианты синдрома Люскан-Люмиш [24]
|
Номер |
Фенотип |
Мутация |
ОНП |
Клинический вариант |
|
.0001 |
СЛЛ |
SETD2, 1-BP DEL, 6341A |
[rs869025569] |
[RCV000208546] |
|
.0002 |
СЛЛ |
SETD2, LEU1815TRP |
[rs869025570] |
[RCV000208561] |
|
.0003 |
СЛЛ |
SETD2, GLN274TER |
[rs869025571] |
[RCV000208536] |
|
.0004 |
СЛЛ |
SETD2, 1-BP DEL, 2028T |
[rs869025572] |
[RCV000208551] |
Примечания: ОНП - однонуклеотидный полиморфизм.
Клинические проявления
Классический фенотип, описанный A. Luscan и соавт. (2014) [7], включает: чрезмерный постнатальный рост; макроцефалию; ожирение; задержку речевого развития; прогрессирующую повышенную оссификацию запястий; удлиненные и крупные конечности. Характерная для СЛЛ краниофациальная дисморфия включает: выступающий большой лоб с высокой линией роста волос; антимонголоидный разрез глаз; длинный нос; длинное лицо; гипоплазию скуловых костей; выступающую нижнюю челюсть (прогнатию). Поведенческие расстройства характеризуются синдромом дефицита внимания с гиперактивностью (СДВГ), конверсионными приступами, агрессивностью, РАС, которые ведут к нарушению обучаемости и трудовой деятельности. Кроме того, при СЛЛ возможны задержка психомоторного развития, интеллектуальный дефицит, тревожное расстройство, генерализованные тонико-клонические припадки, мальформация Арнольда-Киари 1 типа, сирингомиелия [8]. Также H.S. Lumish и соавт. (2015) [8] описали пациента с ранее диагностированным РАС [12], у которого наблюдались задержка развития, афебрильные судороги, дебютировавшие в 4-летнем возрасте, сниженный уровень невербального интеллекта, макроцефалия.
Диагностика
Диагностика СЛЛ базируется на фенотипировании с выделением классических и дополнительных признаков заболевания (таблица 2), а также результатах нейрорадиологических (КТ, МРТ), нейрофизиологических (видеомониторинг ЭЭГ) и нейропсихологических (тестирование уровня интеллекта, внимания, речи и др.) исследований.
Таблица 2
Клинико-лабораторная диагностика синдрома Люскан-Люмиш
|
Признаки |
Характеристика |
|
Классические клинические |
Краниофациальная дисморфия: макроцефалия; выступающий большой лоб с высокой линией роста волос; антимонголоидный разрез глаз; длинный нос; длинное лицо; гипоплазия скуловых костей; выступающая нижняя челюсть. Задержка интеллектуального и речевого развития, расстройство аутического спектра |
|
Дополнительные клинические |
Постнатальный чрезмерный рост, ожирение, удлиненные и крупные конечности, избыточная карпальная оссификация. Задержка психомоторного развития, интеллектуальный дефицит, тревожное расстройство, эпилептические припадки |
|
Нейрорадиологические классические |
Узловые и точечные гиперинтенсивные сигналы в передних отделах лучистого венца и в полуовальном центре, гидроцефалия третьего и боковых желудочков, прогрессирующая макроцефалия |
|
Нейрорадиологические дополнительные |
Мальформация Арнольда-Киари 1 типа, сирингомиелия, фокальная корковая дисплазия |
|
Нейропсихологические |
Нарушения внимания, речи, задержка психомоторного развития, вариабельное снижение невербального интеллекта |
|
Лабораторные (молекулярно-генетические) |
Мутации гена SETD2 на хромосоме 3p21 |
Заключение
СЛЛ является редким и недостаточно изученным клиническим вариантом РАС. В настоящее время найдены немногочисленные описания клинических случаев СЛЛ, а само заболевание включено в OMIM в 2016 году. В связи с этим настороженность практикующих врачей неврологов и психиатров в отношении ранней диагностики СЛЛ низкая. Однако ряд авторов [7; 8; 14] считают целесообразным включение поиска наиболее распространенных мутаций в гене SETD2 в генетическую панель «Расстройства аутистического спектра» при проведении клинического секвенирования экзома у детей с фенотипом РАС, а в случае отсутствия позитивного результата клинического секвенирования экзома – возможно проведение полноэкзомного секвенирования ДНК или прямого секвенирования гена SETD2 по Сенгеру для поиска более редких мутаций, в том числе de novo.
Библиографическая ссылка
Савинова А.В., Шаравии В.Б., Шнайдер Н.А., Насырова Р.Ф. СИНДРОМ ЛЮСКАН-ЛЮМИШ КАК РЕДКОЕ РАССТРОЙСТВО АУТИЧЕСКОГО СПЕКТРА У ДЕТЕЙ ДОШКОЛЬНОГО ВОЗРАСТА // Современные проблемы науки и образования. 2019. № 4. ;URL: https://science-education.ru/ru/article/view?id=28974 (дата обращения: 02.07.2026).