



Спиноцеребеллярная атаксия 1 типа (СЦА 1) (OMIM 164400) и дентаторубропаллидолюисовая атрофия (ДРПЛА) (MIM 125370) относятся к полиглутаминовым заболеваниям. Термин «полиглутаминовые заболевания» в настоящее время применяется для обозначения особой группы тяжелых нейродегенеративных болезней, характеризующихся наличием динамической мутации – так называемой экспансии тринуклеотидных повторов цитозин-аденин-гуанин (CAG) в кодирующих областях соответствующих генов [1; 2]. К полиглутаминовым заболеваниям относятся хорея Гентингтона, шесть форм аутосомно-доминантных спиноцеребеллярных атаксий, дентаторубропаллидолюисова атрофия и спинально-бульбарная амиотрофия Кеннеди. Эти заболевания характеризуются тяжелым прогрессирующим инкурабельным течением, аутосомно-доминантным типом наследования (кроме СБМА, наследуемой по Х-сцепленному рецессивному механизму) и общностью ряда типичных клинико-генетических феноменов, выраженной межсемейной вариабельностью с точки зрения дебюта, сочетания симптомов и даже патоморфологии. Фенотип полиглутаминовых заболеваний является результатом комбинации факторов, таких как мутантный ген, длина экспандированного повтора и длительность болезни [3].
Аутосомно-доминантная спиноцеребеллярная атаксия 1 типа представляет собой нейродегенеративное расстройство, вызванное повторным расширением тринуклеотида CAG в пределах кодирующей области гена Ataxin1 (ATXN1), для которого характерны прогрессирующий тип течения, клинический полиморфизм. Клинические проявления спиноцеребеллярной атаксии 1 типа (СЦА 1) характеризуются развитием мозжечково-пирамидного синдрома, его быстрым прогрессированием, приводящим к социальной дезадаптации больных, к значительному укорочению продолжительности их жизни. СЦА 1 является одним из 5 «якутским» наследственным заболеванием с самой высокой распространенностью среди якутов, чем среди другой популяции мира. Распространенность СЦА 1 за последний 21 год удвоилась на 46 случаев на 100 000 сельских жителей [4; 5].
Дентаторубропаллидолюисовая атрофия является аутосомно-доминантной спиноцеребеллярной атаксией, вызванной экспансией CAG-повторов в гене ATN1, который расположен на хромосоме 12p13 [1]. Патологические повторы CAG достигают 48 или более повторений, а также возраст начала и клинической тяжести заболевания коррелирует с длиной CAG–повтора. Симптоматика дентаторубро-паллидолюисовой атрофии чрезвычайно полиморфна. Заболевание может начинаться в возрасте от 1-го до 6-го десятилетия жизни и проявляется в виде хорсоатетоза, атаксии, деменций, миоклоний, эпилептических припадков [3; 6].
Высокая частота отдельных форм моногенных заболеваний, гомогенность, географическая изолированность и достаточная численность якутской популяции делают ее применимой для проведения молекулярно-генетических исследований по генетическому картированию как моногенных, так и мультифакториальных заболеваний [7-9].
Цель работы: клинико-генеалогический и молекулярно-генетический анализ на наличие мутаций в генах ATXN1 и ATN1 у пациентов с мозжечковым синдромом в Якутии.
Материалы и методы исследования
Для выявления мутации в генах ATXN1 (СЦА 1), ATXN2 (СЦА 2), ATXN3 (СЦА 3), CACNL1A4 (СЦА 6), TBP (СЦА 17) и ATN1 (ДРПЛА) были использованы образцы ДНК 177 пациентов из коллекции биоматериала ЯНЦ КМП и УНУ «Геном Якутии» (рег. № USU_507512). Все больные обращались за консультацией генетика в Медико-генетический центр Республиканской больницы № 1 Национального центра медицины (МГЦ РБ № 1 НЦМ) и в кабинет генетика больницы Якутского научного центра комплексных медицинских проблем (ЯНЦ КМП). Экспериментальная часть работ по поиску экспансии CAG–повторов в генах ATXN1 и ATN1 была проведена в лаборатории наследственной патологии отдела молекулярной генетики ЯНЦ КМП. Все образцы ДНК были получены с информированного согласия исследуемых лиц. Общее количество выборки составило 177 человек (95 женского пола, 82 мужского пола), из них 175 индивидов в возрасте от 20 до 80 лет и 2 ребенка в возрасте 9 и 11 лет, проживающих в Республике Саха (Якутия). Комплексное обследование включало в себя оценку неврологического статуса, электронейрофизиологическое исследование (электроэнцефалография (ЭЭГ)), применение методов нейровизуализации (магнитно-резонансная томография (МРТ), компьютерная томография (КТ)). Молекулярно-генетическая часть исследований мутаций в генах: ATXN1 и ATN1 была проведена на генетическом ДНК-анализаторе (Applied Biosystems 3130). Результаты секвенирования обработаны с помощью программного обеспечения GeneMapper. Стационарное лечение 22 больных проходили на базе больницы ЯНЦ КМП. Статистическая обработка данных проведена с помощью методов описательной статистики.
Результаты и обсуждение. Клинико-генеалогический и молекулярно-генетический анализ на наличие мутаций в гене ATXN1 был проведен среди всей выборки 177 человек. Из них у 104 пациентов с экспансией CAG-повторов в гене ATXN1 выявлен средний возраст начала заболевания в зависимости от количества патологических CAG-повторов. По количеству патологических повторов больные разделены на 5 групп (табл. 1). Как видно из таблицы 1, чем больше количество патологического повтора, тем раньше появляются симптомы заболевания.
Таблица 1
Число CAG-повторов в гене ATXN1 и возраст начала заболевания
|
№ п.гр. |
Количество больных |
Количества патологических CAG-повторов |
Средний возраст начала заболевания |
|
1 |
32 |
39-46 |
46,035± 0,47 |
|
2 |
49 |
46-50 |
35,361± 0,38 |
|
3 |
22 |
51-55 |
34,047± 0,54 |
|
4 |
0 |
56-60 |
|
|
5 |
1 |
>60 |
20 |
На базе больницы ЯНЦ КМП с октября 2017 г. по август 2018 г. прошли стационарное лечение 22 больных из 104 пациентов, с подтвержденным диагнозом СЦА 1. Всем был назначен поддерживающий курс терапии: аминоплазмаль 250,0 через день № 5, сосудистая терапия, ЛФК. Всех больных подвергали оценке по шкале SARA (Scale for the Assessment and Rating of Ataxia). 19 больных хорошо перенесли курс аминокислот и отмечали отчетливое субъективное и объективное улучшение специфических атаксических дисфункций координации движений.
Пример 1. Больной Д., 57 лет. Диагноз: СЦА 1 типа, аутосомно-доминантный тип наследования, прогредиентное прогрессирующее течение. Умеренно выраженный мозжечково-пирамидный синдром. Считает себя больным с 51 года, когда появилась шаткость в походке. Нарушение речи с 52 лет. Дисфагия с 55 лет. Ходит с тростью с 56 лет. Был болен отец, из шести братьев и сестер болеет двое, кроме больного, страдает младшая сестра. Количество CAG-повторов 27/42. Прошел поддерживающую терапию 2 раза с разницей в 5 месяцев. В ходе лечения аминоплазмалем и сосудистой терапией отмечает незначительное улучшение походки в утреннее время, меньше начало шатать, речь стала лучше, особенно по утрам, стал лучше выговаривать слова. По шкале SARA до поддерживающей терапии аминокислотами: походка – 5 баллов, в позе Ромберга - 4 балла, в положении сидя – 2 балла, нарушение речи – 3 балла, тест на дисметрию – 1 балл, пальценосовая проба (ПНП) – 1 балл, адиадохокинез – 2 балла, коленно-пяточная проба (КПП) – 2 балла. Общее количество баллов - 20. SARA после двух курсов поддерживающей терапии: походка – 5 баллов, в позе Ромберга - 4 балла, в положении сидя – 0 баллов, нарушение речи – 1 балл, тест на дисметрию – 1 балл, ПНП – 1 балл, адиадохокинез – 2 балла, коленно-пяточная проба КПП – 1,5 балла. Общее количество баллов – 15,5. Улучшение по шкале тяжести атаксии на 4,5 балла.
Пример 2. Больная П., сестра Д., 52 г. Диагноз: СЦА 1 типа, прогредиентное течение с умеренно выраженными локомоторными нарушениями. Мозжечковый синдром. Хроническая ишемия головного мозга 1-2 ст., субкомпенсация. Шейный, грудной остеохондроз. Цервикокраниалгия. Дебют в 50 лет, появилось нарушение глотания, головокружение. Количество CAG-повторов 27/39. Отмечает улучшение состояние в виде улучшения координации. При оценивании по шкале SARA до поддерживающей терапии аминокислотами: походка – 2 балла, в позе Ромберга - 2 балла, в положении сидя – 1 балл, нарушение речи – 0 баллов, тест на дисметрию – 1 балл, ПНП – 0 балл, адиадохокинез – 1 балла, КПП – 2 балла. Общее количество баллов - 9. SARA после двух курсов поддерживающей терапии: походка – 1 балл, в позе Ромберга - 1 балл, в положении сидя – 0 баллов, нарушение речи – 0 баллов, тест на дисметрию – 0 баллов, ПНП – 0 баллов, адиадохокинез – 0 баллов, КПП – 1 балл. Общее количество баллов – 3. Улучшение по шкале тяжести атаксии на 6 баллов.
Пример 3. Больной Н., 37 лет. Диагноз: СЦА1 типа. Спастический тетрапарез. Выраженный мозжечково-пирамидный синдром. Дизартрия. Заболел с 28 лет, когда появилось нарушение речи и походки. Дисфагия с 32 лет. Ходит с тростью с 35 лет. Ходьба с посторонней помощью с 36 лет. Количество CAG-повторов 30/53. Генеалогический анамнез неизвестен, усыновлен. Был осмотрен в 2012 г., SARA – 12 баллов. В ходе проведенного одного курса поддерживающей терапии улучшения специфической атаксической дисфункции по шкале SARA до и после курса не было. Походка – 5 баллов, в позе Ромберга - 5 баллов, в положении сидя – 2 балла, нарушение речи – 4 балла, тест на дисметрию – 1 балл, ПНП – 2 балла, адиадохокинез – 1 балл, КПП – 2 балла. Общее количество баллов - 22.
Мы предполагаем, что более быстрый и длительный эффект от курса поддерживающей терапии дают пациенты с наименьшим количеством патологического CAG-повтора. Для подтверждения своей теории требуется наблюдение больных в динамике в более большой выборке.
Из 177 пациентов с мозжечковым синдромом у 73 экспансия CAG-повторов в гене ATXN1 не обнаружена. Была проведена ДНК-диагностика на пять форм АД СЦА. Мутация в генах ATXN2 (СЦА 2), ATXN3 (СЦА 3), CACNL1A4 (СЦА 6), TBP (СЦА 17) в обследованной выборке не была выявлена. Обнаружена экспансия CAG-повтора в гене ATN1 (DRPLA) у пятерых больных из одной якутской семьи (рис. 1). У пробанда и у брата 63 мутантных CAG-повторов, у матери и сестры пробанда – 62 (порог нормы ≤ 36), племянник пробанда прошёл досимптоматическое обследование – 19/63 CAG-повторов (табл. 2).
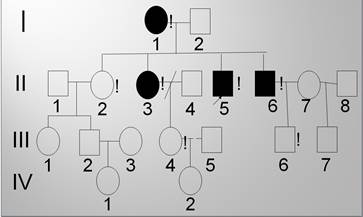
Фрагмент родословной якутской семьи с ДРПЛА. Примечание: I, II, III, IV – нумерация поколений; 1, 2, 3… - нумерация членов семьи в поколении.  – мужской пол,
– мужской пол,  - женский пол,
- женский пол, 
 - больные ДРПЛА, ! – лично обследованные,
- больные ДРПЛА, ! – лично обследованные,  - пробанд
- пробанд
Таблица 2
Клинико-генеалогические и молекулярно-генетические характеристики больных ДРПЛА
|
Больной |
II-5 |
I-1 |
II-3 |
II-6 |
|
Пол |
Мужской |
Женский |
Женский |
Мужской |
|
Возраст |
42 |
66 |
45 |
37 |
|
Возраст манифестации |
36 |
50 |
40 |
36 |
|
Атаксия |
+ |
+ |
+ |
+ |
|
Хореоатетоз |
- |
+ |
+ |
- |
|
Психические нарушения |
Психоорганический синдром |
Апатия |
Дисфория |
_ |
|
Деменция |
- |
+ |
- |
- |
|
Дизартрия |
+ |
+ |
+ |
+ |
|
Гиперкинезы |
+ |
+ |
+ |
- |
|
Глубокие рефлексы с рук и ног |
Живые |
Низкие |
Высокие |
Высокие |
|
Горизонтальный нистагм |
+ |
- |
+ |
+ |
|
SARA |
25 |
33 |
16 |
8 |
|
CAG-повторы |
23/63 |
19/61 |
19/61 |
19/63 |
Как видно из таблицы 2, у четверых членов семьи наблюдается поздний дебют заболевания, в клинике преобладают мозжечково-пирамидный синдром с экстрапирамидной недостаточностью и различной степенью психические нарушения. Мать пробанда монголоидной расы воспитывалась в детском доме Олекминского района, родителей не знает, заболела в 50 лет. Из четверых детей мутация передалась трем детям, возраст начала заболевания после 35 лет, также наблюдается увеличение патологического CAG–повтора на 2 единицы у сыновей. При нейровизуализации отмечались признаки атрофии головного мозга, с преимущественным поражением стволовых структур и мозжечка у всех больных. При проведении ЭЭГ выявлены невыраженные диффузные изменения БЭА головного мозга у сибсов пробанда и умеренная дисфункция подкорково-стволовых структур регистрируются с редкими вспышками эпиактивности по передним отделам головного мозга у пробанда.
Заключение. В данном исследовании был проведен клинико-генеалогический и молекулярно-генетический анализ на наличие мутаций в генах ATXN,1 ATXN2, ATXN3, CACNL1A4, TBP и ATN1 у 177 пациентов с мозжечковым синдромом в Якутии. Выявлено 2 формы полиглутаминовых заболеваний: СЦА 1 типа и ДРПЛА в якутской популяции. Из них у 104 пациентов с экспансией CAG-повторов в гене ATXN1 выявлен средний возраст начала заболевания в зависимости от количества патологического CAG-повтора. Выявлено, что чем больше количество патологического повтора, тем раньше появляются симптомы заболевания. В ходе стационарного наблюдения за 22 больными СЦА 1 мы предполагаем, что более быстрый и длительный эффект от курса поддерживающей терапии дают пациенты с наименьшим количеством патологического CAG-повтора. 19 больных хорошо перенесли курс аминокислот и отмечали отчетливое субъективное и объективное улучшение специфических атаксических дисфункций координации движений. Для подтверждения своей теории требуется наблюдение больных в динамике в более большой выборке.
Экспансия CAG-повтора в гене ATN1 (ДРПЛА) обнаружена у пятерых членов якутской семьи. У пробанда количество мутантных CAG-повторов 63, у матери 61, сибсов 61 и 63. У всех четверых наблюдается поздний дебют и в клинике развитие хореоатетоза, атаксии, психических нарушений, что соответствует данным в изученной литературе о больных ДРПЛА в Японии, Кубе и Латинской Америке.
Аутосомно-доминантные атаксии имеют высокую распространенность во всем мире, они трудно дифференцируются, только молекулярно-генетическим методом можно точно определить тип атаксии. В настоящее время отсутствует этиотропный метод лечения, но если можно улучшить качество жизни, улучшив специфические атаксические дисфункции координации движений, курсом поддерживающей терапии, то актуально создавать неврологические центры с нейрореабилитацией для больных СЦА. Своевременное выявление и направление больных с мозжечковым синдромом в медико-генетическую консультацию позволяет проводить мероприятия по профилактике развития СЦА в отягощенных семьях в Якутии, снижая тем самым «патологический генетический груз» в популяции.
Исследование было проведено в рамках НИР «Изучение генетической структуры и груза наследственной патологии популяций Республики Саха (Якутия)» и профинансировано за счет средств, выделенных для поддержки биоресурсных коллекций (№ 007-03-646 / 2).
Библиографическая ссылка
Варламова М.А., Назарова П.С., Ильинова Е.А., Павлова Н.И., Сидорова О.Г., Кононова С.К., Соловьева Н.А., Дьяконова А.Т., Куртанов Х.А. КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЕ И МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ ОСОБЕННОСТИ У ПАЦИЕНТОВ СО СПИНОЦЕРЕБЕЛЛЯРНОЙ АТАКСИЕЙ 1 ТИПА И ДЕНТАТОРУБРОПАЛЛИДОЛЮИСОВОЙ АТРОФИЕЙ В ЯКУТИИ // Современные проблемы науки и образования. 2018. № 6. ;URL: https://science-education.ru/ru/article/view?id=28147 (дата обращения: 02.07.2026).
DOI: https://doi.org/10.17513/spno.28147