



Малые убиквитин-подобные белки (SUMO) (от англ. Small Ubiquitin – related Modifier) встречаются в клетках эукариот и используются для модификации функциональной активности других белков [6]. Убиквитин является наиболее известным посттрансляционным модификатором белков, однако, помимо убиквитина, существуют другие белки этого семейства, направляющие целевые белки по различным путям в клетке. Такими альтернативными белками-модификаторами у эукариот являются: SUMO (Sentrin, Smt3), NEDD8 (Rub1), ISG15 (UCRP), APG8, APG12, FAT10, Ufm1, URM1 и Hub1.
Белки SUMO эукариот, включая дрожжевой SMT3, имеют трехмерную структуру, сходную со структурой убиквитина, и характеризуются плотно уложенными бета-слоями, обернутыми вокруг единственной альфа-спирали [1]. Эти белки синтезируются в виде различных изоформ белков-предшественников [10].
Протеазы SUMO катализируют два типа реакций – отщепление сигнального белка SUMO от белка–субстрата и формирование изоформ белков-предшественников SUMO. Семейство протеаз SUMO включает в себя два типа в дрожжах (Ulp1 и Ulp2) и шесть типов - у человека (SENP 1-3, SENP 5-7) [11]. Подобно всем представителям семейства протеаз SUMO протеаза SENP2 осуществляет как процессинг незрелых белков SUMO1, SUMO2 и SUMO3 до их зрелых форм, так и отщепление SUMO1, SUMO2 и SUMO3 от белков-субстратов. Кроме того, SENP2 играет активную роль в адипогенезе и промотирует стабилизацию CEBPB [4; 13].
Каталитический домен протеазы SENP2 находится в C-концевой области белка, его длина составляет 165 аминокислотных остатков (а.о.) [база данных UNIPROT Q9HC62]. Связываясь с белком-мишенью, протеазы семейств как Ulp, так и SENP узнают не отдельный сайт, а трехмерную структуру белков SUMO [2; 3; 12].
Способность протеазы SENP2 или ее С-концевого каталитического домена распознавать белок SUMO, находящийся в N-концевой области химерных полипептидов, и осуществлять процессинг химеров с получением целевых зрелых белков используется в специально сконструированных системах экспрессии чужеродных генов in vitro. Распознавание и процессинг не зависят от природы аминокислотного остатка в позиции +1 целевого белка (исключение составляет пролин) относительно С-концевой последовательности Gly-Gly SUMO. В таких системах осуществляют клонирование и экспрессию чужеродных генов, кодирующих целевые белки в виде химерных предшественников, содержащих в N-концевой области последовательность белка SUMO, которая затем удаляется путём протеолиза с помощью протеаз семейства Ulp либо SENP [8; 9]. Эти системы экспрессии используют для: 1) увеличения уровня синтеза чужеродных белков в клетках эукариотических и прокариотических организмов; 2) увеличения растворимости чужеродных белков и обеспечения их секреции; 3) получения зрелых белков с измененными N-концевыми остатками [8].
Задача данной работы заключалась в получении содержащего каталитический домен рекомбинантного С-концевого фрагмента протеазы SENP2 человека, который мог бы быть использован для специфичного выщепления N-концевой последовательности SUMO из состава химерных рекомбинантных белков. Для проверки специфической активности рекомбинантной протеазы был сконструирован и экспрессирован в клетках E. coli ген химерного белка SUMO3-CometGFP.
Материалы и оборудование
Реактивы: акриламид, N,N’-метилен-бисакриламид, додецилсульфат натрия (SDS), персульфат аммония, N, N, N’, N’-тетраметилэтилендиамин (ТЕМЕД), бромфеноловый синий, 2-меркаптоэтанол, этилендиаминтетрауксусная кислота (ЭДТА), канамицин, агароза, изопропил-β-D-1-тиогалактопиранозид (ИПТГ), набор для ПЦР с полимеразой REDTaq корпорации Sigma-Aldrich (США). Дрожжевой экстракт, пептон, агар фирмы Difco (Великобритания). Ni-NTA-сефароза 6В-CL, набор для выделения ДНК из агарозного геля фирмы Qiagen (США). Набор для ПЦР с использованием высокоточной полимеразы Phusion фирмы ThermoFisher Scientific (США). Эндонуклеазы рестрикции, набор для быстрой сшивки ДНК компании «Сибэнзим» (Россия). ТХУ (трихлоруксусная кислота), фенол, хлороформ, этиловый, изоамиловый и изопропиловый спирты, кислоты, щелочи, соли квалификации «ХЧ» или «ОСЧ» производства «Реахим» (Россия).
Штаммы: Escherichia coli: BL 21 (DE3) {F– ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5])}.
Приборы и оборудование: амплификаторы ДНК «БИС 111-02» и «Терцик» (Россия), электропораторы BioRad GenePulserXcell и MicroPulser (USA), твердотельные термостаты «БИС» (Россия). Автоклавы DGM-300 и ВКа-75-П3, ферментёр VortexLab-10, шейкер Heidolph Unimax 2010. Ультразвуковой дезинтегратор МЭФ92 («Мэлфиз», Россия). Центрифуги: J2-21 и Allegra X-12R фирмы Beckman; Eppendorf 5418.
Питательные среды и их компоненты: среда LB (Lysogeny Broth, пептон 1%, дрожжевой экстракт 0,5%, NaCl 1%) и низкосолевая среда LB (Lysogeny Broth, пептон 1%, дрожжевой экстракт 0,5%, NaCl 0.5%). Фосфатно-солевой буфер pH 7.4 (137 mM NaCl 2.7 mM KCl) («БиолоТ», Россия).
Методы
Получение штамма E. coli – продуцента протеазы SENP2. Нуклеотидную последовательность фрагмента гена, кодирующего С-концевую область протеазы SENP2, включающую её каталитический регион, проектировали с использованием компьютерной программы GeneDesigner 2.0 фирмы DNA2.0 (США), обеспечивающей оптимизацию кодонов в гене для эффективной его экспрессии в клетках E. coli. Кроме того, с применением разработанной нами программы проводили поиск пар кодонов, затрудняющих, по данным [5], трансляцию мРНК в E. coli, и осуществляли их замену на синонимические кодоны.
Синтезированный ген был клонирован в клетках E. coli BL 21 (DE3) в составе экспрессирующей плазмиды pJExpress401 (DNA2.0, США), обеспечивающей индуцируемую экспрессию гена под контролем промотора бактериофага Т5. С помощью ПЦР отбирали колонии трансформантов, выросшие на чашках с агаризованной средой LB, содержащей 30 мкг/мл канамицина.
Наработка и очистка рекомбинантной протеазы SENP2. Отобранные колонии трансформированных клеток E. coli BL 21 (DE3) анализировали на способность осуществлять индуцируемый ИПТГ синтез полипептидов, соответствующих по молекулярной массе ожидаемому размеру целевого фрагмента протеазы SENP2. Отобранный клон-продуцент выращивали в 500 мл свежей среды LB, содержащей 30 мкг/мл канамицина, до достижения оптической плотности (600 нм) = 0,9. Затем в растущую культуру вносили IPTG до концентрации 1 mM и продолжали культивирование в течение 12 часов при 30 °С на орбитальном шейкере со скоростью 200 об/мин.
Клетки собирали центрифугированием и суспендировали в 8 мл лизирующего буфера: 50 мМ Трис-HCl, рН 8,0; 500 мМ NaCl; 6 М гуанидин гидрохлорид; 20 мМ имидазол рН 8,0. Суспензию клеток озвучивали во льду 6 раз по 30 сек с перерывами по 1 мин при помощи ультразвукового дезинтегратора. Лизат центрифугировали в течение 15 мин при 3000 xg.
Колонку, содержащую 2 мл Ni-NTA агарозы (Qiagen), уравновешивали лизирующим буфером. Смолу инкубировали с осветлённым лизатом в течение часа при медленном переворачивании. Смолу промывали двумя объёмами по 4 мл денатурирующего буфера «А» (8 M мочевины; 50 mM Трис-HCl; 500 mM NaCl) с pH 8,0, двумя объёмами по 4 мл буфера «А» с pH 6,0 и 4 мл буфера «А» с pH 5,3. Смолу переносили на колонку и поводили элюцию буфером «А» с pH 4.0. В полученный элюат вносили Твин-20 до концентрации 0,1%. Ренатурацию фермента проводили, последовательно диализуя элюат против буфера «Б» (50 мМ Трис-HCl рН 8,0; 300 мМ NaCl; 2 мМ дитиотреитола и 0,1% Твин-20), содержащего постепенно снижающуюся концентрацию мочевины от 6 до 0 М.
Получение штамма E. coli-продуцента химерного белка SUMO3-CometGFP.
Ген зелёного флюоресцирующего белка CometGFP амплифицировали методом ПЦР в реакционной смеси, содержащей в качестве матрицы плазмидную ДНК pEcometGFP (DNA 2.0 USA), праймеры 405F GTAGTCTCAAGGTATGACGGCATTGACGGAAG и 406R GTCTCTAGATTAACGGTAAGTTTCCAGGTCGAC и высокоточную ДНК-полимеразу Phusion. Полученный ампликон гидролизовали рестриктазами BsmAI и XbaI и встраивали в плазмиду pE-SUMO3-Kan (LifeSensors USA) по сайту BsaI с помощью ДНК-лигазы фага Т4 [7]. Лигазной смесью трансформировали клетки E. coli BL21(DE3). Клоны, содержащие рекомбинантную плазмиду pE-SUMO3-CometGFP, отбирали на чашках с низкосолевой средой LB с антибиотиком зеоцином 50 мкг/мл.
Наработка и очистка белка-субстрата SUMO3-CometGFP
Отобранный клон, содержащий плазмиду pE-SUMO3-CometGFP, растили в 500 мл низкосолевой среды LB, содержащей 50 мкг/мл зеоцина, при 37 °С на орбитальном шейкере (200 об/мин) до достижения оптической плотности при 600 нм = 0,8. Затем в культуру вносили IPTG до концентрации 1 mM и продолжали выращивание 4 часа при 37 °С на орбитальном шейкере (200 об/мин).
Клетки собирали центрифугированием (3500 xg), суспендировали в 8 мл буфера «Б» (Tris HCl 20 mM, pH 8,0; NaCl 150 mM), вносили 8 мг лизоцима и инкубировали 30 мин при 0 °С. Лизат клеток получали озвучиванием клеток 7 раз по 3 мин при помощи ультразвукового дезинтегратора. Лизат осветляли центрифугированием при 3000 xg в течение 15 мин и инкубировали со смолой Ni-NTA, предварительно насыщенной ионами никеля и уравновешенной буфером «Б». Смолу наносили на колонку и проводили промывку буфером «Б» с 20 mM имидазола. Элюцию белка проводили буфером «Б», содержащим 250 mM имидазола.
Определение концентраций ДНК и белков проводили при помощи наборов Qubit dsDNA HS Assay kit Q32854 и Protein Assay Kit Q33212 с использованием флюориметра Qubit Q32857 (ThermoFisher Scientific, США).
Протеолиз белка субстрата SUMO3-CometGFP
Протеолиз белка проводили при 30 °С в различных массовых соотношениях протеаза/субстрат, при pH 7.0. На реакцию брали 30 мкг белка SUMO3-CometGFP в объёме 50 мкл. Пробы по 5 мкл отбирали через 12-24 часа и анализировали электрофорезом в SDS-ПААГ.
Результаты и обсуждение
При проектировании структуры гена фрагмента протеазы SENP2 мы исходили из имеющихся в базе данных GenBank сведений о локализации каталитического центра протеазы SENP2 в С-концевой области фермента. Размер нативной протеазы SENP2 составляет 589 а.о. (GenBank ID: AAG15309; AAH40609; AK027599; BC040609), каталитический центр образуют аминокислотные остатки фермента с 396 по 560 а.о. Чтобы быть уверенными, что полученный нами рекомбинантный фрагмент протеазы SENP2 будет проявлять протеолитическую активность, мы спроектировали рекомбинантный ген, кодирующий фрагмент протеазы SENP2, включающий аминокислотные остатки с 353 по 582 а.о., т.е. существенно перекрывающий область каталитического центра.
Сконструированный и синтезированный ген фрагмента протеазы SENP2 был клонирован в составе экспрессирующего вектора pJexpres401 в клетках E. coli шт. BL21(DE3). Один из отобранных клонов, продуцирующий индуцируемый ИПТГ белок с размерами, соответствующими ожидаемому размеру рекомбинантного фрагмента протеазы SENP2, был использован для препаративной наработки этого фермента в 1 л среды ЛБ. Синтезированный рекомбинантный белок был локализован в тельцах включения бактериальных клеток. Поскольку в С-концевой области рекомбинантной протеазы нами был предусмотрен 6-гистидиновый тракт, то для её очистки проводили аффинную хроматографию на колонке с Ni-NTA сефарозой 6B-CL в денатурирующих условиях, как описано в «методах». На рис. 1 представлены результаты электрофоретического анализа фракций, полученных после аффинной хроматографии лизата индуцированных ИПТГ клеток отобранного клона E. coli str. BL21(DE3), несущего плазмиду pJexpres401/SENP2 fragment.
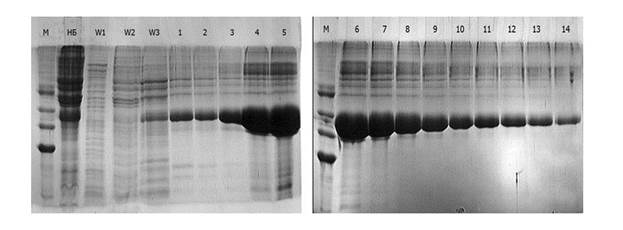
Рис. 1. Электрофореграмма фракций, полученных после аффинной хроматографии лизата индуцированных ИПТГ клеток E. coli str. BL21(DE3), несущих плазмиду pJexpres401/SENP2 fragment. Дорожки: М - маркёр молекулярных масс (38, 29, 22 и 11 кДа); НБ – белки, не связавшиеся со смолой; W1 – белки, смываемые денатурирующим буфером «A» с pH 8,0; W2 - белки, смываемые денатурирующим буфером «A» pH 6,0; W3 – белки, смываемые денатурирующим буфером «A» с pH 5.3. 1-14 – фракции, полученные при элюции белков буфером «А» с pH 4.0
Как можно видеть из рис. 1, с помощью аффинной хроматографии можно добиться ~70-75%-ной очистки рекомбинантного белка, который по своим размерам (~26 кДа) соответствует ожидаемому размеру фрагмента протеазы SENP2.
Для определения протеолитической активности полученного фрагмента протеазы SENP2 была осуществлена генно-инженерная сборка гена, кодирующего химерный полипептид SUMO3-CometGFP, который предполагалось использовать в качестве специфического субстрата для протеазы SENP2.
Ген, кодирующий химерный полипептид SUMO3-CometGFP, его клонирование и проведение наработки и очистки белка-субстрата SUMO3-CometGFP осуществляли как описано в «методах». Поскольку полипептид SUMO3-CometGFP накапливался в цитоплазме бактериальных клеток в растворимом виде, то для её очистки проводили аффинную хроматографию лизата клеток продуцента на колонке с Ni-NTA сефарозой 6B-CL в нативных условиях, как описано в «методах». На рис. 2 представлены результаты электрофоретического анализа фракций, полученных после аффинной хроматографии лизата индуцированных ИПТГ клеток отобранного клона E. coli str. BL21(DE3), несущего плазмиду pE-SUMO3-CometGFP.
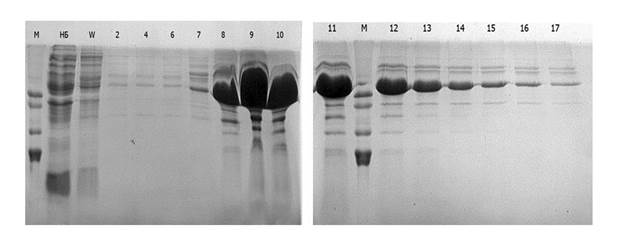
Рис. 2. Электрофореграмма фракций, полученных после аффинной хроматографии лизата индуцированных ИПТГ клеток E. coli str. BL21(DE3), несущих плазмиду pE-SUMO3-CometGFP. Электрофорез фракций проводили в денатурирующих условиях в пластинах 12%-ного полиакриламидного геля. Дорожки: М - маркёр молекулярных масс (38, 29, 22 и 11 кДа); НБ – белки, не связавшиеся со смолой; W – белки, смываемые буфером «Б» с pH 8.0, содержащим 20 mM имидазола. 2-17 – фракции, полученные при элюции буфером «Б» с pH 8.0, содержащим 250 mM имидазола
Как можно видеть из рис. 2, основная масса рекомбинантного химерного полипептида SUMO3-CometGFP обнаруживается во фракциях с 8-й по 14-ю. Белок в этих фракциях по своим размерам (~40 кДа) соответствует ожидаемому размеру полипептида SUMO3-CometGFP. Препарат рекомбинантного полипептида SUMO3-CometGFP имел 85-90%-ную степень очистки.
Химерный полипептид SUMO3-CometGFP был использован в качестве специфического субстрата для определения протеолитической активности полученного нами рекомбинантного фрагмента протеазы SENP2. На рис. 3 представлена электрофореграмма продуктов гидролиза белка-субстрата SUMO3-CometGFP фрагментом протеазы SENP2. Молекулярные массы белков: SUMO3-CometGFP - ~ 39,8 кДа, SUMO3 - 12,5 кДа, CometGFP – 27,3 кДа. В результате проведённого анализа было установлено, что полный гидролиз химера (более 90% белка SUMO3-CometGFP) наблюдается при инкубации белка с рекомбинантным фрагментом протеазы SENP2 в соотношении 1/25 течение 3 суток при 30 °С.
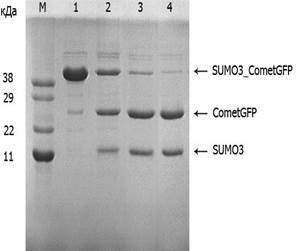
Рис. 3. Электрофореграмма продуктов гидролиза химерного белка–субстрата SUMO3- CometGFP рекомбинантным фрагментом протеазы SENP2. Электрофорез образцов проводили в денатурирующих условиях в пластинах 12%-ного полиакриламидного геля. Дорожки: (1) - исходный белок-субстрат SUMO3-CometGFP, (2-4) - продукты гидролиза белка–субстрата, полученные через 24 (2), 48 (3) и 84 часа (4)
Заключение
С помощью генно-инженерных методов нами были получены препараты двух рекомбинантных белков: С-концевого фрагмента протеазы SENP2, содержащего каталитический центр фермента, и химерного полипептида SUMO3-CometGFP, предназначенного для использования в качестве субстрата для оценки протеолитической активности рекомбинантного фрагмента протеазы SENP2. Продемонстрирована протеолитическая активность фрагмента протеазы SENP2. Однако, по литературным данным, коммерческие препараты SUMO протеаз работают быстрее и имеют более высокую активность. Мы, к сожалению, не имели возможности сравнить полученный нами фермент с коммерческим аналогом из-за сложностей с соблюдением температурного режима при его транспортировке из США. По-видимому, по этой причине коммерческая протеаза SENP2, полученная из США, не проявляла заметной протеолитической активности. Тем не менее в дальнейшем мы планируем с помощью соответствующих праймеров получить укороченные варианты клонированного в настоящей работе фрагмента гена SENP2 и исследовать протеолитические активности кодируемых ими полипептидов.
Библиографическая ссылка
Беклемишев А.Б., Мамаев А.Л. ПОЛУЧЕНИЕ БАКТЕРИАЛЬНОГО ПРОДУЦЕНТА РЕКОМБИНАНТНОЙ ПРОТЕАЗЫ SENP2 // Современные проблемы науки и образования. 2017. № 1. ;URL: https://science-education.ru/ru/article/view?id=26103 (дата обращения: 27.06.2026).