



Исследования и фундаментальные достижения последних десятилетий в микробиологии, генетике и молекулярной биологии дали возможность изучения генетического разнообразия бактериальных штаммов. С развитием генетической систематики, с расширением круга используемых методов, направленных на изучение бактериального генома, и с накоплением экспериментальных данных современная таксономия бактерий стала быстро развиваться, что позволило решить многие спорные вопросы систематики конкретных групп микроорганизмов. Вместе с тем оставались нерешенными проблемы, связанные с противоречиями между данными молекулярной систематики и традиционными представлениями, основанными на анализе фенотипа в отношении группы молочнокислых бактерий, имеющих большое практическое значение.
До недавнего времени при отборе штаммов для создания бактериальных заквасок использовались только стандартные микробиологические подходы оценки стартерных культур, такие как выделение, идентификация таксономического положения на основе изучения их морфологических, физиолого-биохимических свойств, определение условий культивирования и их технологических свойств. Однако использование только этих традиционных технологий не всегда позволяет эффективно отбирать безопасные и технологичные штаммы молочнокислых бактерий для использования их в качестве заквасочных культур в производстве кисломолочных продуктов и пробиотических препаратов. Многими исследователями отмечалось, что у микробиологов возникают трудности при идентификации на видовом и дифференциации на внутривидовом уровне бактерий различных родов факультативных анаэробов. Особенно трудно идентифицировать виды внутри родов: Enterococcus, Lactococcus, Leuconostoc, Pediococcus, Streptococcus, Lactobacillus с помощью классических фенотипических методов. В этой связи особую актуальность приобретает проблема использования современных фундаментальных научных достижений изучения бактериального генетического разнообразия в прикладных областях науки. Фенотипическая характеристика штамма, применяемая для идентификации, паспортизации и типирования штаммов бактерий, в настоящее время уже не является достаточной для установления таксономического положения изучаемого организма и целостной характеристики его свойств. Что же касается штаммов молочнокислых бактерий, практически использующихся в пищевой промышленности, использование современных молекулярно-биологических подходов для таксономической идентификации, молекулярно-генетической паспортизации, генотипирование бактерий позволит выбирать лучшие стартовые культуры для промышленного применения и получения качественных и безопасных пищевых продуктов и бактериальных препаратов.
Лактобактерии являются представителями естественной микрофлоры организма человека и животных и играют важную роль в поддержании колонизационной резистентности [1; 2]. Они подавляют рост и размножение поступающих извне представителей посторонней микрофлоры, предотвращают их приживление. В организме человека лактобациллы появляются в первые дни после рождения и в течение всей жизни присутствуют в кишечнике, препятствуя развитию гнилостных и патогенных микроорганизмов. Развитие дисбактериоза в большинстве случаев связано с нарушением естественного состава микрофлоры кишечника. Представители рода Lactobacillus нашли свое применение в составе многочисленных лечебно-профилактических кисломолочных продуктов и фармакопейных биопрепаратов [3; 4].
Лактобактерии и продукты их метаболизма широко используются для профилактики и лечения различных острых и хронических заболеваний пищеварительного тракта, способствуя восстановлению нормальной микрофлоры. Лактобактерии широко используются в пищевой промышленности в составе заквасок для приготовления кисломолочных продуктов (сыры, масла, йогурты), при хлебопечении (ржаной хлеб), квашении овощей и засолке рыбы, при приготовлении сухих и варено-копченых колбас [5]. Прежде чем использовать штаммы микроорганизмов для промышленного производства, необходимо определить их разнообразные характеристики, в том числе и генетические, которые, в отличие от культурально-морфологических, более стабильны. Классическими методами внутривидового типирования микроорганизмов являются изучение морфологии, биохимические тесты и серологические реакции. Но вышеуказанные методы обладают существенными недостатками: они опираются на выявление вариабельных фенотипических признаков, относительная однородность которых определяет трудности во внутривидовой дифференциации микроорганизмов и не позволяет выявить различия между штаммами одного вида. Для повышения достоверности результатов при типировании штаммов лактобацилл целесообразно наряду с классическими использовать молекулярно-генетические методы исследования [6-8].
Молекулярно-генетические методы исследования позволяют не только провести внутриродовую дифференциацию лактобацилл на основании особенностей их геномов, но и дают возможность получить индивидуальные генотипические характеристики каждого штамма. Наиболее перспективными методами типирования штаммов лактобацилл являются методы полимеразной цепной реакции (ПЦР). Одновременное применение нескольких методов ПЦР обеспечивает возможность генотипирования лактобацилл и позволяет решать задачи дифференциации видов и штаммов, входящих в состав пробиотических и производственных заквасок [9; 10].
Материалы и методы
Определение и анализ нуклеотидных последовательностей генов 16S rRNA
Геномную ДНК выделяли методом Kate Wilson [11]. Концентрацию ДНК измеряли спектрофотометрическим методом с использованием спектрофотометра Nano Drop 1000 при длине волны 260 нм, а также проводили качественную оценку ДНК электрофоретическим методом. Матрицы для секвенирования синтезировали с помощью ПЦР, используя универсальные праймеры 8f-5’–AgAgTTTgATCCTggCTCAg-3 и 806R-5’- ggACTACCAgggTATCTAAT-3 [12], что позволяло амплифицировать ген 16S rRNA почти полностью. Реакционная смесь (30 мкл) содержала 3 мкл 10 х реакционного буфера (Fermentas), 2,5 мM MgCl2 , 0,2 мM каждого дезоксирибонуклеозидтрифосфата (дНТФ), по 10 пмоль каждого из праймеров, 1 единицу Taq –полимеразы Maxima Hot Start Taq DNA Polymerase (Fermentas, США) и 150 нг геномной ДНК в качестве матрицы. ПЦР проводили в термоциклере Mastercycler pro S (Eppendorf). Реакцию начинали инкубированием смеси при 95 °С в течение 7 минут, затем следовало 30 циклов, состоящих из инкубаций: 95 °С – 30 секунд, 55 °С - 40 секунд, 72 °С – 1 мин. Завершающую элонгацию проводили при 72 °С в течение 10 минут. Продукты амплификации разделяли в 1,5%-ном агарозном геле. Гели окрашивали этидиум бромидом. Электрофорез проводили в камере для горизонтального электрофореза Bio-RAD Basic и источником тока Consort EV-243. В качестве электродного буфера использовали 1хТАЕ-буфер. Документирование полученных результатов проводили, используя систему документаций гелей Gel Doc. Размеры молекул анализируемых образцов ДНК определяли путем сопоставления их электрофоретической подвижности маркеров - фрагмент ДНК известной молекулярной массы. В качестве маркера молекулярных масс использовали O'GeneRuler™ 100bp Plus DNA Ladder производства (Fermentas). ПЦР продукты очищали от остатков олигонуклеотидов методом дефосфолирирования с помощью щелочной фосфатазы (SAP-shimp alkaline phosphatase) и эндонуклеазы I [13]. Секвенирование фрагментов гена 16S rRNA идентифицируемых бактерий проводили на автоматическом секвенаторе 3730 х l DNA Analyzer (Applide Biosystems, США) с использованием набора Big Dye Terminator v3.1 Cycle Sequencing Kit (Applide Biosystems, США), придерживаясь рекомендаций производителя. Результаты секвенирования обрабатывали в программе SeqMan (Applide Biosystems). Поиск гомологичных нуклеотидных последовательностей генов 16S rRNA осуществляли с помощью программы BLAST в базе данных Gene Bank Национального центра биотехнологической информации США. Идентификация была осуществлена относительно инвентарных номеров Gene Bank первых трех нуклеотидных последовательностей, имеющих максимальное совпадение. Филогенетический анализ проводили с использованием программного обеспечения MEGA4 [14]. Выравнивание нуклеотидных последовательностей проводили, используя алгоритм ClustalW [15]. Для построения филогенетических деревьев использовали метод «объединения соседей» Neiighbor-Joining (NJ) [16].
Результаты и обсуждение
Филогенетический анализ нуклеотидных последовательностей генов 16S rRNA у рода Lactobacillus
В данном исследовании представлены результаты молекулярно-генетической идентификации штамма молочнокислых бактерий на основе анализа нуклеотидных последовательностей 16S rRNA гена, выделенного из желудочно-кишечного тракта здоровых людей. Штамм молочнокислых бактерий был просеквенирован и проанализирован.
Генетическая идентификация штамма была осуществлена методом определения прямой нуклеотидной последовательности фрагмента 16S rRNA гена, с последующим сравнением нуклеотидной идентичности с последовательностями, депонированными в международной базе данных Gene Bank, а также построением филогенетического дерева с нуклеотидными последовательностями референтных штаммов.
Для точного определения таксономической принадлежности исследуемого штамма был использован метод идентификации с применением видоспецифических праймеров.
Результаты ПЦР-реакций с использованием праймеров приведены на рис. 1.
- LU-5 и RhaII, специфичных для вида Lactobacillus rhamnosus;
- LU-5 и Lpar-4, специфичных для вида Lactobacillus paracasei.
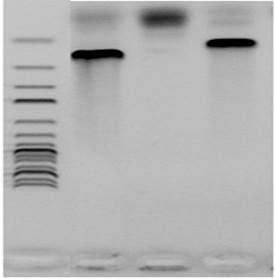
Рис. 1. Результаты ПЦР-анализа
1. Маркер O’GeneRuler 100 bp DNA Ladder (100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1200, 1700, 2000, 3000 п.н., сверху вниз).
2. ПЦР-анализ исследуемого штамма с использованием праймеров LU-5 и Lac2.
3. ПЦР-анализ исследуемого штамма с использованием праймеров LU-5 и RhaII.
4. ПЦР-анализ исследуемого штамма с использованием праймеров LU-5 и Lpar-4.
Первичный скрининг по базе данных Gene Bank и RDP-II показал, что исследуемый штамм принадлежит к следующим систематическим группам: Bacteria; Firmicutes; Lactobacillales; Lactobacillaceae; Lactobacillus, причем гомология с некоторыми видами рода Lactobacillus составляет 97%.
Наработка фрагмента размером 312 п.н. при использовании видоспецифических праймеров LU-5 и Lpar-4 позволяет утверждать, что исследуемый штамм относится к виду Lactobacillus paracasei. Отсутствие фрагмента размером 113 п.н. при использовании видоспецифических праймеров LU-5 и RhaII показывает, что исследуемый штамм не относится к виду Lactobacillus rhamnosus.
Последовательности были выровнены с соответствующими последовательностями ближайших видов бактерий, доступными из базы данных Gene Bank. Результаты обработки секвенсов при помощи компьютерной программы, находящейся на сайте RDB II (Ribosomal Database Project II), предназначенной для определения родства микроорганизмов и построения филогенетических деревьев, представлены в графическом виде.
S000003746 0.977 0.748 1404 Lactobacillus rhamnosus (T); JCM 1136; D16552
S000005601 0.926 0.609 1468 Lactobacillus perolens (T); L532; Y19167
S000009879 0.980 0.813 1431 Lactobacillus zeae (T); ATCC15820; D86516
S000319968 0.983 0.920 1416 Lactobacillus paracasei (T); NBRC 15906; AB181950
S000379822 0.949 0.687 1442 Lactobacillus concavus (T); AS 1.5017; AY683322
S000393517 0.946 0.641 1455 Lactobacillus pantheris (T); AF413523
S000395052 0.977 0.803 1432 Lactobacillus casei (T); ATCC 393; AF469172
S000413948 0.986 0.922 1438 Lactobacillus paracasei (T); JCM 8130; D79212
S000414171 0.950 0.636 1371 Lactobacillus dextrinicus (T); JCM 5887; D87679
S000652804 0.949 0.641 1424 Lactobacillus camelliae (T); MCH3-1; AB257864
Дальнейший анализ по RDP II 16S рРНК базе данных показал гомологию с теми же видами бактерий. Результаты филогенетического анализа последовательностей гена 16S rRNA у изучаемого штамма представлены на филогенетическом дереве (рис. 2), построенном в программе MEGA 4, с использованием Neiighbor-Joining кластерного метода расчета генетических расстояний и bootstrap-анализа, отражающего достоверность кластеризации.
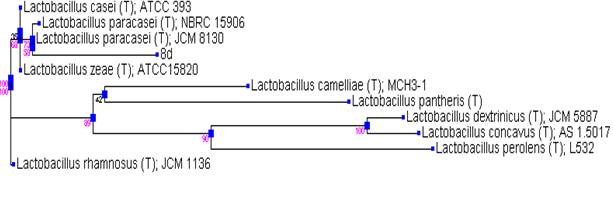
Рис. 2. Филогенетическое дерево, основанное на анализе структур фрагментов гена 16S rRNA, отражающее родственные связи штаммов молочнокислых бактерий рода Lactobacillus
Так как критерием отнесения микроорганизма к тому или иному виду считается гомология не менее 97%, то исследуемый штамм можно отнести к нескольким видам рода Lactobacillus. Сравнение нуклеотидных последовательностей генов 16SrRNA коллекционного штамма с таковой международных баз данных также позволило установить, что тестируемый штамм наиболее близок к виду Lactobacillus paracasei (98%).
Секвенирование гена 16S rRNA также выявило высокий уровень гомологии с представителями рода Lactobacillus и показало их идентичность с референтным штаммом.
Библиографическая ссылка
Кузнецова Т.В., Кебекбаева К.М., Джакибаева Г.Т., Молжигитова А.Е. ГЕНОТИПИРОВАНИЕ ШТАММА МОЛОЧНОКИСЛЫХ БАКТЕРИЙ МЕТОДОМ ПЦР-АНАЛИЗА // Современные проблемы науки и образования. 2016. № 5. ;URL: https://science-education.ru/ru/article/view?id=25349 (дата обращения: 06.06.2026).