



Синдром Шерешевского – Тернера (СШТ) является хорошо изученным хромосомным синдромом с частотой – 1:2000-2500 новорожденных девочек [5,6,26]. СШТ характеризуется низкорослостью, короткой шеей с «крыловидными» складками, нарушением полового развития в виде аплазии или гипоплазии яичников и матки, незначительно развитых вторичных половых признаков, лимфотическим отёком кистей и стоп при рождении, бочкообразной грудной клеткой. Среди МАР могут наблюдаться антимонголоидный разрез и гипертелоризм глазных щелей, эпикант, птоз, низкий рост волос на затылке, высокий и широкий лоб, ретрогения, деформированные низко расположенные ушные раковины, гипертелоризм сосков, вальгусное положение локтевых суставов, гипоплазия ногтей и сосков [6,12,25,30]. Из пороков внутренних органов часто отмечаются аномалии сердца (около 50 %) и почек (около 40 %) [29]. Среди всех хромосомных синдромов СШТ характеризуется большим разнообразием кариотипов, в том числе мозаичных форм, объединенных одним признаком – отсутствием или нарушением структуры хромосомы Х [17]. При мозаичных формах, которые оцениваются по данным литературы в 30–56 % всех случаев СШТ [15,16,29], всегда присутствует клеточный клон 45,Х. Известна высокая степень внутриутробной гибели эмбрионов, имеющих кариотип 45,Х, до 99 % случаев [25]. Среди девочек с низким ростом и задержкой полового развития, проходящих цитогенетическое исследование, СШТ выявляется приблизительно в 25 % [9]. Полиморфизм клинических признаков при СШТ может быть связан с различным кариотипом (мозаицизм c наличием нормального клона клеток, маркерные и кольцевые хромосомы Х) [29]. Присутствие нормальной клеточной линии 46,ХХ может облегчать клинические проявления у пациентов с СШТ [2,6,27,29]. Целью исследования являлась оценка доли регулярных и мозаичный форм синдрома, проведение возможной корреляции генотип-фенотип при разных формах хромосомной аномалии.
Материалы и методы
Ретроспективный анализ результатов цитогенетических и молекулярно-цитогенетических исследований был проведен 96 девочкам с клиническими признаками СШТ. Цитогенетический анализ проводили на хромосомах лимфоцитов периферической крови, культивированных стандартным методом с применением GTG и CBG окрашивания [6]. У каждого пациента было проанализировано не менее 20, а в случае мозаицизма – не менее 30 метафазных пластинок. Молекулярно-цитогенетические исследования проводили методом флюоресцентной гибридизации in situ (FISH) для уточнения доли мозаичного клона, определения генетической природы маркерных и кольцевых хромосом, уточнения сложных структурных перестроек. В случаях мозаицизма проводился анализ 50–100 метафазных пластинок и 1000 интерфазных ядер [31]. В работе использовались оригинальные ДНК пробы из коллекции лаборатории цитогенетики и геномики психических заболеваний ФГБНУ «Научного центра психического здоровья». Описание этих ДНК проб представлено в работах проф. Ю.Б. Юрова с соавт. [35,36] и д-ра И.В. Соловьева с соавт. [11,28]. Применяли ДНК пробы MCB (Multicolor Chromosome Banding), окрашивающие полосы хромосомы в разные цвета [24]. При анализе мозаицизма клон 45,Х учитывался в том случае, если он был выше 2,3 %, т.е. выше значения, определенного для нормальных девочек возрастом до 18 лет [18]. Оценка клинических особенностей пациентов с СШТ проводилась по историям болезни. Запись хромосомных аномалий проводилась согласно международной номенклатуре по цитогенетике человека (ISCN, 2016).
Результаты и обсуждение
В работе представлены результаты цитогенетических и молекулярно-цитогенетических исследований 96 девочек. Средний возраст девочек составлял 10,5+4,1 лет (от 0,3 до 17 лет). Удельный вес различных кариотипов при СШТ представлен в таблице 1. При этом синдроме принято выделять такие кариотипы, как 45,Х, мозаицизм 45,Х/46,ХХ (45,X/47,XXX/46,XX), наличие в кариотипе изохромосомы Х по длинному плечу - i(Xq), кольцевые и маркерные хромосомы Х, материал хромосомы Y в кариотипе, делеции хромосомы Х [29].
Таблица 1
Различные кариотипы у обследованных пациентов с СШТ
|
кариотип |
число случаев |
% от всех случаев СШТ |
|
45,X |
27 |
28,1 |
|
45,X/46,XX(45,X/47,XXX/46,XX) |
36 |
37,5 |
|
46,X,i(Xq) |
1 |
1,04 |
|
46,X,i(Xq)/45,X |
6 |
6,25 |
|
46,X,i(Xq)/45,X/46,XX |
2 |
2,1 |
|
46,X,idic(X)(q22.2)/45,X |
1 |
1,04 |
|
46,X,r(X)/45,X |
8 |
8,3 |
|
46,X,mar(derX)/45,X |
6 |
6,25 |
|
46,X,mar(derY)/45,X |
3 |
3,1 |
|
46,X,del(Yq) |
1 |
1,04 |
|
46,X,der(X)t(Xp;Yq) |
2 |
2,1 |
|
45,X/45,X,der(20)t(20p;Yq) |
1 |
1,04 |
|
46,X,del(Xp) |
1 |
1,04 |
|
46,X,del(Xp)/45,X |
1 |
1,04 |
Кариотип 45,Х был обнаружен у 27 девочек (28,1 %). Помимо основных клинических признаков синдрома, в некоторых случаях наблюдалась тугоухость, гипертрихоз, пигментные невусы. У четырех девочек была отмечена задержка психоречевого и/или психомоторного развития (ЗПРР, ЗПМР). В одном случае симптомокомплекс был особенно тяжелым: ЗПРР, ЗПМР, микроцефалия, судороги и МАР в виде гипоплазии нижней челюсти, оттопыренных ушных раковин. Существует гипотеза о том, что практически все пациентки с СШТ имеют мозаицизм с присутствием клона нормальных клеток, а нежизнеспособны эмбрионы с регулярной формой 45,Х [1,5,6,20]. Данные последних исследований указывают на то, что потеря хромосомы Х у рожденных девочек с СШТ обусловлена в первую очередь митотическими факторами, и клон 45,Х возникает на стадии первых дроблений зиготы с нормальным кариотипом. Кроме того, известен ген PSF2RA, расположенный в псевдоаутосомных участках коротких плеч хромосом Х и Y, необходимый для нормального функционирования плаценты, и, вероятно, девочки, рожденные с кариотипом 45,Х, жизнеспособны благодаря тому, что клетки плаценты в этих случаях имели нормальный кариотип и были способны осуществлять трофическую функцию [20]. Для выявления возможного мозаицизма было проведено FISH исследование 9 девочкам с кариотипом 45,Х, в результате которого клон 46,ХХ, составлявший в среднем 3,2 % клеток, был обнаружен у троих пациентов, что подтверждало данные литературы [2,23,34].
Наиболее многочисленной группой в нашем исследовании были девочки с мозаичным кариотипом 45,X/46,XX или 45,X/47,XXX/46,XX, составлявшие 37,5 % от всех пациентов с СШТ. Доля клона 45,Х варьировала у разных пациентов от 5 % до 95 % и в среднем составляла 30,3 %. Явной корреляции генотип-фенотип в зависимости от доли аномального клона клеток в этой группе не наблюдалось. Так, например, девочки с долей клеток 45,Х в пределах 5 % имели низкий рост, первичную аменорею, гипоплазию и аплазию внутренних половых органов, аномалии почек точно также, как и девочки, у которых доля клона 45,Х составляла более 50 %. У девочки с 6 % клеток 45,Х помимо выраженных признаков СШТ наблюдались черты аутизма и снижение интеллекта. Другая девочка с долей клеток 45,Х, равной 13 %, имела грубые ЗПРР и ЗПМР, эпилепсию. Вероятно, это связано с долей аномальных клеток в различных тканях и органах (головной мозг, почки, сердце и т.д.). Мозаицизм 45,X/47,XXX/46,XX после цитогенетического исследования был выявлен у 5 девочек, однако, после проведения FISH анализа девочкам с мозаицизмом 45,Х/46,ХХ, дополнительный клеточный клон 47,ХХХ выявлялся примерно в половине случаев, но представлен был в небольшой доле клеток (1–5 %) (рис.1 а,б,в). Исключение представлял случай кариотипа 47,ХХX/45,Х/47,XX,+r(X)/46,XX с процентным соотношением клонов: 67%/27%/2%/4% у девочки 14 лет с низким ростом, задержкой полового развития, гипертелоризмом глазных щелей и сосков, широкой грудной клеткой и сандалевидной щелью стоп. В этом случае клон 47,ХХX был преобладающей клеточной линией, однако клинические признаки СШТ не отличались от обычных. Мозаичной формы 45,Х/47,ХХХ среди наших случаев обнаружено не было. Учитывая отсутствие явной корреляции клинических проявлений с долей обнаруженных аномальных клеток лимфоцитов, можно предположить о существовании тканевого мозаицизма, в частности, затрагивающего органы и ткани, в которых содержание клеток с кариотипом 45,Х может быть значительно выше, чем в клетках крови. Несмотря на это, для установления диагноза СШТ важен факт выявления аномального клона в клетках различных тканей, включая и лимфоциты периферической крови.
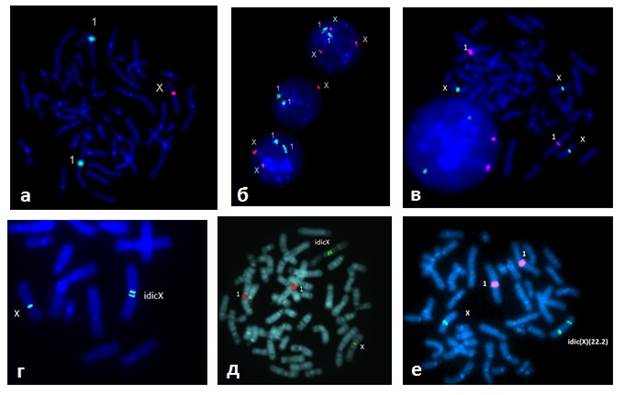
Рис. 1. Результаты FISH исследования с центромерным ДНК зондом на хромосому Х. (для контроля плоидности при интерфазном анализе применялся ДНК зонд на хромосому 1): а) метафазная пластина – 45,Х; б) интерфазные ядра с тремя, одной и двумя хромосомами Х (сверху вниз) у одного пациента; в) метафазная пластина – 47,ХХХ; г) нормальная и изодицентрическая хромосома Х – idic(X)(p11.2) (табл.2, №9); д) позднореплицирующаяся изодицентрическая хромосома Х, специфическая окраска после культивирования с BrDU указывает на инактивацию перестроенной хромосомы (табл.2, №8); е) нормальная и изодицентрическая хромосома Х с точкой разрыва Хq22.2. (табл.2, №10)
Присутствие изохромосомы Х по длинному плечу – i(Xq) в кариотипе наблюдалось в 9 случаях и в одном – изохромосома с точкой разрыва в длинном плече q22.2. Общее число случаев с изохромосомой Х составило 10,4 % (табл. 2). Все случаи были мозаичными за исключением одного, в котором выполнялось только цитогенетическое исследование, в связи с чем невозможно утверждать, что и в этом случае не было мозаицизма. В семи случаях клон 46,X,i(Xq) сочетался с клоном 45,Х, в двух – присутствовал клон нормальных клеток.
Таблица 2
Анализ случаев с изохромосомой Х
|
№ |
Кариотип |
Процентное соотношение клонов |
|
46,Х,i(X)(q10) |
- |
|
|
46,Х,i(X)(q10)/45,X/46,XX |
50/30/20 |
|
|
46,Х,i(X)(q10)/45,X/46,ХХ |
20/37/43 |
|
|
46,Х,i(X)(q10)/45,X |
92/8 |
|
|
46,Х,i(X)(q10)/45,X |
7/93 |
|
|
46,Х,i(X)(q10)/45,X |
62/38 |
|
|
46,Х,i(X)(q10)/45,X |
88/12 |
|
|
46,Х,idic(X)(p11.2)/45,X |
76/24 |
|
|
46,X,idic(X)(р11.2)/45,X |
35/65 |
|
|
46,X,idic(X)(q22.2)/45,X |
94/6 |
В двух случаях изохромосома Х была дицентрическая с точкой разрыва в коротком плече р11.2, в одном – в длинном плече idic(X)(q22.2) (рис. 1 г,д,е). В последнем случае изохромосома была необычной, поскольку состояла из части длинных плеч и из двух коротких плеч хромосомы Х. У 16-летней девушки с этим кариотипом была отмечена незрелость внутренних половых органов, низкого роста не отмечалось. У остальных девочек симптомокомплекс соответствовал СШТ, однако не отмечалось отека кистей и стоп при рождении. У двух девочек (табл.2, №2 и 6) 6 и 7 лет, соответственно, наблюдались ЗПРР и ЗПМР. По данным литературы, в случаях с перестроенной хромосомой Х в кариотипе аномальная хромосома Х подвергается инактивации в большинстве клетках [6,22]. Одной девочке (табл. 2, № 8) был проведен анализ особенности инактивации хромосом Х, показавший, что изохромосома Х была инактивирована в 100 % клеток, что соответствовало литературным данным (рис.1 д).
Кольцевые хромосомы Х (n=8) и маркерные хромосомы, производные от хромосомы Х (n=6), выявлялись в 14,6 % (табл. 3).
Таблица 3
Анализ кольцевых и маркерных хромосом Х при СШТ
|
№ |
кариотип |
Процентное соотношение клонов |
|
45,Х/46,X,r(X)/46,XX |
64/7/9 |
|
|
45,Х/46,X,r(X)(p21q21) |
50/50 |
|
|
45,Х/46,X,r(X)(p22q21) |
40/60 |
|
|
45,Х/46,X,r(X)/46,XX |
64/32/4 |
|
|
45,Х/46,X,r(X)(p11.2q21) |
55/45 |
|
|
45,Х/46,X,r(X)/46,X,der(X) |
71/28/1 |
|
|
45,X/46,X,r(X)(p11.2q12)/ 46,X,idic(X)(p11.2)/ 47,X,idic(X)(p11.2),idic(X)(p11.2) |
48/43/8/1 |
|
|
45,X/46,X,r(X)(p11.22q13.1) |
22/78 |
|
|
45,Х/46,X,der(X) |
30/70 |
|
|
45,Х/46,X,der(X) |
85/15 |
|
|
45,Х/46,X,der(X) |
50/50 |
|
|
45,Х/46,X,der(X) |
92/8 |
|
|
45,Х/46,Х,der(X) |
38/62 |
|
|
45,Х/47,ХХ,+der(X)/46,XX |
26/58/16 |
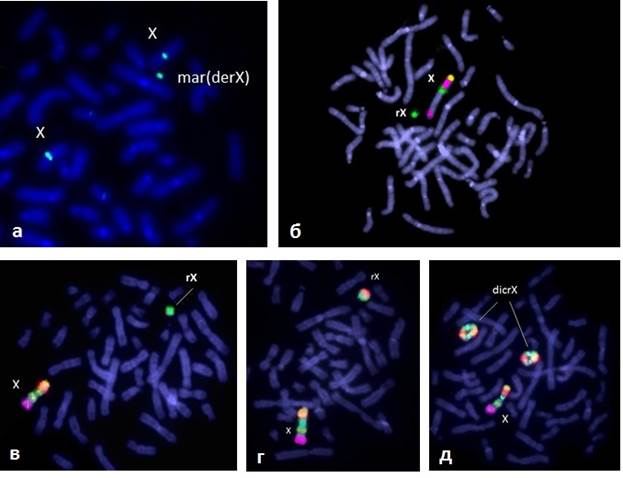
Рис. 2. FISH и MCB диагностика маркерных и кольцевых хромосом Х: а) маркерная хромосома Х (табл. 3, №11), центромерный ДНК зонд; б) кольцевая хромосома Х (табл.3, №7), МСВ проба; в) кольцевая хромосома Х (табл.3, №5), МСВ проба; г,д) кольцевая и две дицентрические кольцевые хромосомы Х в метафазной пластине (табл. 3, № 3), МСВ проба
Эти случаи рассматриваются вместе, т.к. кольцевая хромосома небольшого размера при цитогенетическом исследовании часто фиксируется, как маркерная хромосома (рис.2 а,б,в). Во всех этих случаях присутствовал дополнительный клон клеток 45,Х, в трех случаях – клон нормальных клеток. В некоторых случаях кольцевые хромосомы сочетались с минихромосомами (derX) (табл. 3, № 6) и с изодицентрическими хромосомами Х (табл. 3, №7). Происхождение маркерных и кольцевых хромосом определяли методом FISH с использованием центромерных ДНК проб во всех случаях. Точки разрыва в кольцевых хромосомах определяли при использовании сайтспецифичных ДНК проб или MCB пробы (multicolor chromosome banding). Клинические проявления в этой группе хромосомных аномалий соответствовали СШТ, однако в двух случаях (табл. 3, № 7 и 9) отмечались ЗПРР, ЗПМР, а в одном (табл. 3, № 8) – у девочки 1,5 лет с кольцевой хромосомой r(X)(p11.22q13.1) – задержка внутриутробного развития в сочетании с ЗПМР, микроцефалией, атрезией хоан и МАР: высоким нёбом, поперечной бороздой ладоней, низко расположенными ушными раковинами. При анализе генотип-фенотип этой группы девочек было отмечено, что наиболее тяжелые клинические особенности, включающие ЗПРР, ЗПМР, наблюдаются при кольцевых хромосомах, не имеющих центр инактивации ХIST, располагающемся в участке Xq13.2, а также при маркерных хромосомах (дериватных минихромосомах Х), содержащих эухроматиновые участки. В случаях кольцевых хромосом с геном ХIST и маркерных хромосом Х, состоящих из прицентромерного гетерохроматина без эухроматиновых участков, у пациентов отмечали только признаки СШТ (рис.2 а, в, г, д). Такие же клинические особенности были характерны для мозаицизма с небольшим клоном, содержащим дериватную хромосому Х.
Материал хромосомы Y был выявлен в кариотипе у 7 девочек (7,3 %) (табл. 4), среди которых маркерная минихромосома Y в сочетании с клоном 45,Х – в двух случаях (табл. 4, № 1 и 2), немозаичный случай делеции длинного плеча хромосомы Y (табл. 4, № 3), изодицентрическая хромосома Y по короткому плечу в сочетании с клоном 45,Х (табл. 4, № 4), два случая несбалансированной транслокации с участием длинного плеча хромосомы Y и короткого плеча хромосомы Х (табл. 4, № 5 и 6) и один необычный случай (табл. 4, № 7) мозаичной несбалансированной транслокации хромосомы Y c хромосомой 20 при регулярной моносомии хромосомы Х.
Таблица 4
Анализ материала хромосомы Y в кариотипе у девочек с СШТ
|
№ |
Кариотип |
Процентное соотношение клонов |
|
45,Х/46,X,der(Y) |
70/30 |
|
|
45,X/46,X,der(Y) |
57/43 |
|
|
46,X,del(Y)(q11.21) |
- |
|
|
46,X,idic(Y)(q11.22)/45,X |
89/11 |
|
|
46,X,der(X)t(X;Y)(p22.3;q11.2) |
- |
|
|
46,X,der(X)t(X;Y)(p22.31;q11.22) |
- |
|
|
45,Х/45,X,der(20)t(Y;20)(q11.2;p13) |
27/73 |
При обследовании пациентов с СШТ случаи присутствия материала хромосомы Y в кариотипе всегда рассматриваются отдельно в связи с большим риском возникновения у таких девочек гонадобластомы [19,23,25]. Все случаи были уточнены методом FISH с применением различных ДНК проб. В клиническом плане все девочки имели симптомокомплекс, характерный для СШТ. У одной девочки (табл. 4, № 6 и рис. 3) наблюдалась ЗПРР. В этом случае при несбалансированной транслокации между хромосомами Х и Y имелась частичная моносомия дистального участка хромосомы Х. Интерес представляет случай (табл. 4, № 4), при котором преобладающим клоном клеток был клон с изодицентрической хромосомой Y (около 90 %) с двумя копиями гена SRY в двух коротких плечах изохромосомы, однако, несмотря на это, наблюдался женский фенотип. В этом случае можно предполагать тканевой мозаицизм. Также интересен случай № 7 в табл.4, когда в части клеток материал хромосомы Y был расположен на хромосоме 20, тогда как в других клетках гомологичные хромосомы 20 были нормальными и хромосома Y в них не выявлялась. При этом девочка имела только признаки СШТ без умственной отсталости. Ни одна девочка с материалом хромосомы Y не имела гонадобластому. Возраст этих девочек был от 6 до 16 лет (средний возраст 12,9+3,7 лет).
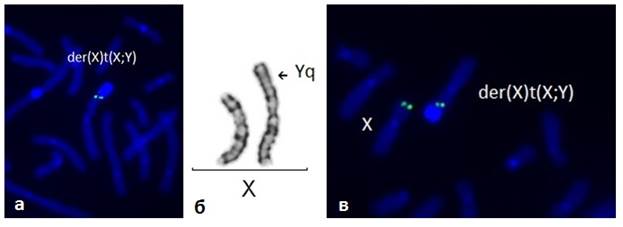
Рис. 3. Дериватная хромосома Х – результат транслокации хромосом Х и Y (табл. 4, случай № 6), кариотип 46,X,der(X)t(X;Y)(p22.31;q11.22): а) FISH с использованием ДНК зонда на участок Yq11.22, который присутствует в аномальной хромосоме; б) нормальная и дериватная хромосомы Х (GTG-окраска); в) FISH с использованием ДНК зонда на участок Хр22.31, присутствующего в аномальной хромосоме Х
Делеции короткого плеча хромосомы Х были отмечены у двух девочек, что составило 1,1 %, обнаруженные цитогенетически на хромосомах высокого разрешения и уточненные методом FISH [10]. В одном случае терминальная делеция короткого плеча хромосомы Х выявлена при мозаицизме 45,Х[82]/46,X,del(X)(p21) [18], в другом обнаружена регулярная интерстициальная делеция – 46,X,del(X)(p22.1p22.1). У первой девочки были классические признаки СШТ, у второй – гипоплазия внутренних половых органов, однако рост был нормальный, вероятно в связи с тем, что ген SHOX, расположенный в терминальной части короткого плеча хромосомы Х, не был поврежден.
Заключение
Исследование показало, что доля мозаичных форм кариотипа была велика и составила 67,7 %. Мозаицизм с присутствием нормального клона 46,ХХ был обнаружен в 39,6 % случаев. Процент выявленных мозаичных форм в наших исследованиях оказался больше значений, указанных в литературе (30–56 %) [15,16,29], что, вероятно, было возможно благодаря применению FISH метода при анализе большого числа клеток у каждого пациента [3,4]. Анализ корреляции генотип-фенотип при мозаичных формах показал, что классический фенотип СШТ проявлялся у пациентов как при большой доле аномальных клеток, так и при доле, составлявшей 5–10 %, что может свидетельствовать о возможном наличии тканевого мозаицизма [1,6,8,9,14,34]. При мозаичных формах у пациентов часто наблюдали пигментные невусы, а также асимметрию тела и лица. Пигментные пятна отмечались и у девочек с регулярным кариотипом 45,Х, что свидетельствует о возможном «скрытом» тканевом мозаицизме. Более тяжелые клинические проявления, включающие ЗПРР, ЗПМР, аутизм, микроцефалию, судороги, наблюдались нами у 13 девочек (13,5 %) с различными формами кариотипа, что также можно связать с тканевым мозаицизмом (большая доля аномальных клеток в головном мозге). Возможно, в случаях кольцевых и дериватных хромосом Х с отсутствием центра инактивации XIST тяжелые состояния могут объясняться транскрипционной активностью генов в аномальной хромосоме Х. Нельзя исключать и другие, сочетанные с СШТ, геномные нарушения. Вероятно, для определения истинных причин тяжелых клинических форм при СШТ, а также для анализа корреляций фенотип-генотип, необходимы другие методы исследования, такие как микроматричный хромосомный анализ (array CGH, SNP array) [7,13,32,33] или экзомное секвенирование. Таким образом, различные кариотипы при СШТ предполагают индивидуальный подход в каждом случае и необходимость совместного применения цитогенетического и молекулярно-цитогенетических (FISH, MCB, array CGH) исследований, а также исследование различных тканей.
Библиографическая ссылка
Колотий А.Д., Ворсанова С.Г., Юров Ю.Б., Соловьев И.В., Демидова И.А., Кравец В.С., Шаронин В.О., Куринная О.С., Гордеева М.Л., Богатырева Е.П., Юров И.Ю. ЦИТОГЕНЕТИЧЕСКИЕ И МОЛЕКУЛЯРНО-ЦИТОГЕНЕТИЧЕСКИЕ (FISH И MCB ) ИССЛЕДОВАНИЯ СИНДРОМА ШЕРЕШЕВСКОГО-ТЕРНЕРА: РЕТРОСПЕКТИВНЫЙ АНАЛИЗ 96 СЛУЧАЕВ // Современные проблемы науки и образования. 2016. № 5. ;URL: https://science-education.ru/ru/article/view?id=25121 (дата обращения: 29.06.2026).