



Генетическая инженерия конкретнее и точнее клеточной по характеристике используемых объектов и оперирует в основном с разными по форме и размерам фрагментами клетки [1; 2]. Термины «генетическая инженерия», «генная инженерия» и «рекомбинантная ДНК» - равноценны [3].
Генетическую инженерию можно представить как соединение фрагментов ДНК природного и синтетического происхождения или их комбинацию in vitro с последующим введением полученных рекомбинантных структур в живую клетку для того, чтобы введенный фрагмент ДНК после включения его в хромосому либо реплицировался, либо автономно экспрессировался.
Успехи генетической инженерии привели к тому, что свыше 100 белков человека (биорегуляторов, корректоров гомеостаза, факторов врожденного и приобретенного иммунитета) могут сохранять свою видеоспецифичность. Они нарабатываются как лекарственные средства путем микробиологического синтеза. При этом технология рекомбинантной ДНК позволяет их совершенствовать: повышать физиологическую активность, снижать вероятность побочных реакций после введения и так далее [4].
Основным при получении рекомбинантных белков является решение проблемы дефицита сырья, так как из человеческих тканей в промышленном масштабе получать их невозможно. В качестве продуцентов рекомбинантных белков человека чаще других в настоящее время используются: E.coli (кишечная палочка), Bacillus subtilis (сенная палочка), Saccharomyces cerevisiae (пекарские дрожжи). Эти организмы достаточно безопасны, однако попадание их в окружающую среду по ряду причин нежелательно. В связи с этим существуют принятые и тщательно соблюдаемые правила работы с рекомбинантами [5]. Безопасность должна соблюдаться на генетическом и на физическом уровне, и это относится к производству любых рекомбинантных белков.
Данная работа направлена на разработку генноинженерного инструментария, позволяющего получать рекомбинантный фермент аминогидролазу, востребованную в пищевой и биотехнологической промышленности, с использованием штамма - суперпродуцента Escherichia coli.
Целью данной работы являлось: получение новой рекомбинантной аминогидролазы Pseudomonas putida и характеристика ее биохимических свойств.
Объекты и методы исследований
Для исследования использован коллекционный штамм Pseudomonas putida B5437, предоставленный ФГУП «ГосНИИгенетика». Для культивирования Pseudomonas putida B5437 использовали LB-среду, содержащую 0,1% (NH4)2SO4, 1 mM MgSO4, 1,05% K2HPO4, 0,45% KH2PO4, 0,4% сукцината и 0,2% глюкозы.
Определение нуклеотидной последовательности проводилось методом секвенирования на геномном секвенаторе GS Junior (Roche, Япония).
Клонирование гена hutI проводили методом полимеразной цепной реакции (ПЦР). Разработку олигонуклеотидных праймеров для амплификации гена проводили с помощью программы OLIGO (версия 3.3) с учетом данных о первичной структуре гена hutI. В качестве матрицы для амплификации, кодирующей область гена hutI из P. Putida, использовали последовательность данного гена из баз данных GenBank (X12702.1). Для наработки к-ДНК методом полимеразной цепной реакции использовали соответствующие праймеры:
прямой праймер ‒ CTCGAGATGAAACTTGTCTTCCTCG;
обратный праймер ‒ GAATTCAATGGTGATGGTGATGGTG.
Полимеразную цепную реакцию проводили в 20–50 мкл раствора, приготовленного на основе десятикратного буфера для Taq-полимеразы, который содержал 200 мкМ каждого из дезоксинуклеозидтрифосфатов, 0,5 мкМ праймеров, 2 мМ MgSO4, 10 нг матрицы, 2 единицы Taq ДНК-полимеразы и 0,1 единицы Pfu ДНК-полимеразы. Температуру отжига олигонуклеотидов рассчитывали по эмпирической формуле Tm = 67,5+34[%GC]–395/n, где %GC = (G+C)/n, n – число нуклеотидов. Анализ продуктов ПЦР проводили электрофорезом в 1%-ном геле агарозы.
Для экспрессии целевого белка в работе использованы клетки штамма E. coli BL21[DE3]Star (Invitrogen, США) с генотипом F-ompT hsdSB (rB-mB-) gal dcm rne131 (DE3), содержащие в геноме λDe3 лизоген и мутацию rne131. Мутированный ген rne (rne131) кодирует усеченную форму РНК-азы Е, что уменьшает внутриклеточное разрушение м-РНК, приводя к увеличению ее ферментативной стабильности.
Для получения эффективной системы экспрессии гена hutI вектор pET28а+ модифицировали последовательностью гена hutI. Гидролиз фрагментов ДНК рестриктазами XhoI u HindIII проводили в объеме 100 мкл в специальных буферных растворах при оптимальной для каждого фермента температуре 37 °С. Полноту гидролиза контролировали с помощью гель-электрофореза в 1%-ном агарозном геле, содержащем 0,5 мкг/мл бромистого этидия.
Предварительно подготавливали компетентные клетки E. coli BL21DE3 к трансформации. Для этого брали отдельную колонию клеток, выращенную на LB-агаре, и помещали в 5 мл LB-среды. Клетки культивировали в течение ночи при 37 °С и постоянном перемешивании (250 об/мин); 2 мл полученной ночной культуры помещали в 200 мл LB-среды. Клетки растили при 37 °С при постоянном перемешивании (250 об/мин) до ОП600 = 0,6, после чего осаждали центрифугированием в течение 10 мин на 4000 g при +4 °С. Клетки отмывали в деионизованной воде в исходном объеме с последующим центрифугированием. Процедура отмывки повторялась трижды. После отмывки осадок клеток ресуспендировали в малом объеме деионизованной воды и центрифугировали 30 с при 5000 об/мин на микроцентрифуге. К осадку клеток добавляли 3 объема (от объема клеточного осадка) 15%-ного раствора глицерина, осадок ресуспендировали и быстро замораживали в жидком азоте. Готовые к трансформации клетки хранили при –70 °С.
Трансформацию компетентных клеток E. coli BL21DE3 осуществляли методом электропорации. Для этого 2 мкл плазмидной ДНК с концентрацией 0,3–1 нг/мкл добавляли к 12 мкл компетентных клеток, перемешивали и проводили электропорацию на генераторе высоковольтных импульсов ГВИ-1 в стерильных ячейках при электрическом импульсе напряженностью 10 кВ/см длительностью 4 мсек. После трансформации клетки помещали в 1 мл SОС-cреды (2% бактотриптон, 0,5% дрожжевой экстракт, 10 мМ NaCl, 2,5 мМ KCl, 10 мМ MgCl2, 10 мМ MgSO4, 20 мМ глюкозы) и инкубировали в течение 40 мин при 37 °С. После инкубации 10–250 мкл клеточной суспензии высеивали на селективную LB-среду, содержащую канамицин (25 мкг/мл), для отбора рекомбинантных клонов.
Для автоиндукции экспрессии по методу Штудиера использовалась модифицированная среда PYP-5052, состоящая из 1% пептона, 0,5% дрожжевого экстракта, 50 мМ Na2HPO4, 50 мМ K2HPO4, 25 мМ (NH4)2SO4, 2 мМ MgSO4, 0,5% глицерина, 0,05% глюкозы и 0,2% лактозы. В среду PYP-5052, содержащую канамицин в концентрации 25 мкг/мл, инокулировали единичную колонию штамма-продуцента. После этого ферментировали при 25 или 37 °С в термостатированном шейкере роторного типа при 250 об/мин в течение 32 или 18 ч или до отсутствия существенного изменения ОП600 за 1 ч. Далее отбирали аликвоту клеток на анализ. Аликвоты хранили при –20 °С.
Экспрессию рекомбинантного фермента контролировали с использованием электрофореза в полиакриламидном геле по Лэммли. Ферментацию проводили в 2-литровых колбах в 250 мл среды, содержащей канамицин в концентрации 25 мкг/мл, в термостатированной качалке роторного типа при 37 °С и 250 об/мин.
Определение содержания белка имидазолонпропионат-аминогидролазы в клетках E. coli методом денситометрического анализа полученной электрофореграммы с использованием компьютерной программы TotalLab.
Для определения стабильности очищенного препарата рекомбинантной имидазолонпропионат-аминогидролазы его разбавляли в 50 раз в таких же буферах с рН от 3 до 12 и концентрацией 0,1 М. Концентрация белка в пробах составляла 0,2 мг/мл. Образцы инкубировали при +30 °С в течение 10 мин. Затем отбирали аликвоты и вносили их в инкубационную смесь для определения активности в стандартных оптимальных условиях при рН 8,5.
Результаты и их обсуждение
С помощью программы SignalP 3.0 и базы данных NCBI [6] идентифицирован ген hutI, кодирующий имидазолонпропионат-аминогидролазу.
Праймеры содержали на 5’-концах дополнительные последовательности, включающие сайты рестрикции NcoI для прямого праймера и HindIII для обратного, и были предназначены для амплификации и последующей встройки структурной области гена в полилинкер экспрессирующего вектора pET28a по соответствующим сайтам. Обратный праймер был сконструирован таким образом, чтобы полученный ампликон не содержал стоп-кодона и обеспечивалась состыковка рамок считывания гена и последовательности His6.
В результате клонирования гена hutI всего получено шесть клонов, содержащих 5,0-kb фрагменты.
Для создания системы экспрессии гена, кодирующего имидазолонпропионат-аминогидролазу, использован коммерческий вектор pET28a+, содержащий в своем составе ключевые позиции, которые отражены в таблице 1.
Таблица 1
Описание участков рЕТ28а+
|
Участок |
Позиция |
|
T7 promoter |
370-386 |
|
T7 transcription start |
369 |
|
His•Tag coding sequence |
270-287 |
|
T7•Tag coding sequence |
207-239 |
|
Multiple cloning sites |
|
|
(BamH I - Xho I) |
158-203 |
|
His•Tag coding sequence |
140-157 |
|
T7 terminator |
26-72 |
|
lacI coding sequence |
773-1852 |
|
pBR322 origin |
3286 |
|
Kan coding sequence |
3995-4807 |
|
f1 origin |
4903-5358 |
Физическая карта экспрессионного вектора представлена на рисунке 1.
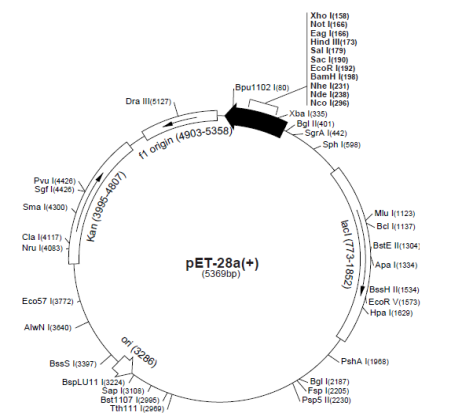
Рис. 1. Коммерческий экспрессионный вектор pET28а+
Экспрессию рекомбинантного фермента контролировали с использованием электрофореза в полиакриламидном геле по Лэммли. На рисунке 2 представлены результаты электрофореза.
Был оптимизирован уровень экспрессии имидазолонпропионат-аминогидролазы по времени и температуре индукции. В качестве индуктора использовали ИПТГ в концентрации 1 мМ. Индукцию экспрессии проводили, когда ОП600 культуры клеток достигла 0,6–0,8 оптических единиц. После этого через определенное время (от 15 мин до 1 ч) из культур клеток после индукции отбирали аликвоты для анализа. В качестве контроля использовали культуры клеток без индукции. Без индукции клетки растут по логарифмическому закону: до 0,6–0,8 оп.ед. наблюдается рост по линейной зависимости, далее до 3,8–4,2 оп.ед. происходит экспоненциальный рост, после чего наблюдается выход кривой роста на плато в стационарную фазу роста. После добавления к культуре клеток индуктора 1 мМ ИПТГ наблюдается замедление роста клеток, изменение количества клеток с 0,6 до 1,5 оп.ед. происходит за 4 ч, однако далее наблюдается начало экспоненциального роста культуры клеток с более поздним выходом на плато в стационарную фазу роста.
При изучении динамики биосинтеза рекомбинантного белка имидазолонпропионат-аминогидролазы клетками E. coli после индукции экспрессии 1 мМ ИПТГ было показано, что максимальный уровень экспрессии гена hutI в клетках E. coli при индукции 1 мМ ИПТГ наблюдается после 3-часовой ферментации. При этом, по данным денситометрического анализа, выход белка составляет 32% от общего клеточного белка (рис. 2). При проведении индукции экспрессии при температуре 25 °С наблюдался более длительный выход культур клеток в экспоненциальный рост, составивший при температуре индукции 25 °С 9 часов.

Рис. 2. Анализ лизатов трансформированных клеток E. coli BL21DE3 после индукции экспрессии в различных условиях:
1 – маркер молекулярного веса PageRuler™ Prestained Pro-tein Ladder (Fermentas); 2 – отрицательный контроль (лизат клеток штамма-продуцента без добавления индуктора проведения индукции); 3 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 0,2%-ной лактозой по методу Штудиера при температуре 25 °С в течение 32 ч; 4 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 0,2%-ной лактозой по методу Штудиера при температуре 37 °С в течение 18 ч; 5 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 1 мМ ИПТГ при температуре 37 °С в течение 3 ч; 6 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 1 мМ ИПТГ при температуре 37 °С в течение 5 ч; 7 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 1 мМ ИПТГ при температуре 25 °С в течение 5 ч; 8 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 1 мМ ИПТГ при температуре 25 °С в течение 8 ч; 9 – лизат трансформированных клеток E. coli BL21DE3 после индукции экспрессии 1 мМ ИПТГ при температуре 25 °С в течение 3 ч.
При этом максимальный уровень экспрессии (32%) для всех температур индукции оставался неизменным.
Результаты изучения индукции экспрессии 0,2%-ной лактозой по методу Штудиера представлены на рисунках 3, 4.
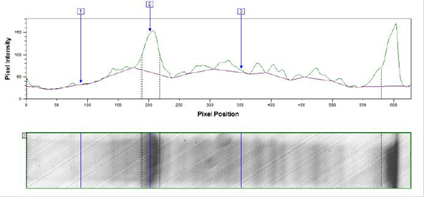
Рис. 3. Денситограмма лизата трансформированных клеток E. coli BL21DE3 после индукции экспрессии 0,2%-ной лактозой по методу Штудиера при температуре 25 °С в течение 32 ч.
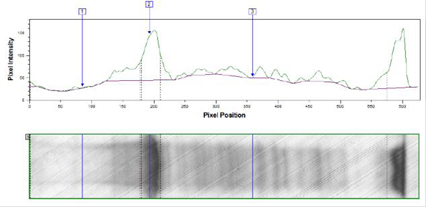
Рис. 4. Денситограмма лизата трансформированных клеток E. coli BL21DE3 после индукции экспрессии 0,2%-ной лактозой по методу Штудиера при температуре 37 °С в течение 18 ч.
Средняя оптическая плотность (ОП600) культуры трансформированных клеток E. coli BL21DE3 была одинаковой при разных температурах культивирования и составила 7 оп.ед.
В результате денситометрического анализа было показано, что в трансформированных клетках E. coli BL21DE3 изучаемый белок накапливается в количестве 35% от общего клеточного белка при температуре ферментации 37 °С и 33% при температуре 25 °С.
Данный метод индукции экспрессии был выбран как более простая, эффективная и недорогая альтернатива классической индукции с использованием ИПТГ в системах экспрессии, основанных на лактозном опероне. При использовании автоиндукции не требуется ни следить за оптической плотностью культуры клеток, ни добавлять индуктор.
Разработанная схема очистки рекомбинантного фермента включает дробное осаждение сульфатом аммония и две хроматографические стадии: металлохелатную и гидрофобную.
Для анализа чистоты фермента использовали диск-электрофорез в полиакриламидном геле (ПААГ) с додецилсульфатом натрия. Результаты очистки представлены в таблице 2.
Таблица 2
Результаты очистки рекомбинантной имидазолонпропионат-аминогидролазы Pseudomonas putida по оптимизированной схеме
|
Стадия очистки |
Экстракт |
Осаждение сульфатом аммония (25-50%) |
Ni-NTA-сефароза |
Гидрофобная хроматография |
Гидрофобная хроматография, «хвост» |
|
Удельная активность, Е/мг |
0,276 |
0,822 |
2,217 |
3,316 |
2,228 |
|
Активность, Е/мл |
2,57 |
16,60 |
11,64 |
12,80 |
4,10 |
|
Белок, мг/мл |
9,30 |
20,20 |
5,25 |
3,90 |
1,80 |
|
Объем, мл |
110,0 |
16,5 |
20,0 |
13,7 |
10,5 |
|
Суммарная активность, Е |
282,7 |
273,9 |
232,8 |
175,4 |
43,1 |
|
Суммарный белок, мг |
1023,0 |
333,0 |
105,0 |
52,9 |
19,3 |
|
Выход, % |
100 |
97 |
85 |
58 |
14 |
Данные, представленные в таблице 2, свидетельствуют о том, что при гидрофобной хроматографии фермент элюируется двумя пиками. При этом 80% фермента элюируется в первой фракции с удельной активностью 3,3 Е/мг, а около 20% фермента с удельной активностью 2,2 Е/мг элюируется во второй фракции. Выход фермента в результате очистки составляет около 60%.
Определяли стабильность очищенного препарата рекомбинантной имидазолонпропионат-аминогидролазы.
Полученные результаты представлены на рисунке 5.
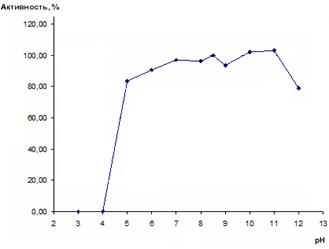
Рис. 5. Стабильность препарата рекомбинантной имидазолонпропионат-аминогидролазы при различных значениях рН
Из рисунка 5 следует, что рекомбинантная имидазолонпропионат-аминогидролаза наиболее стабильна при рН 7,0 и более щелочных значениях вплоть до рН 11,0. При рН 11,0 даже происходит некоторое увеличение активности. По-видимому, это объясняется тем, что ингибирование фермента ДТТ является обратимым и происходит быстрее при рН 11,0. Кроме того, в кислой среде (рН ниже 7,0) белок быстро и необратимо инактивируется. Например, при рН 5,0 сохраняется 83% активности, а при рН 4,0 фермент уже полностью инактивируется после 10-минутной инкубации при +30 °С.
Зависимость активности рекомбинантной имидазолонпропионат-аминогидролазы от температуры определяли в стандартной реакционной смеси, которую предварительно выдерживали 10 мин при температуре реакции. Измерения проводили на спектрофотометре с термостатом, поддерживающим нужную температуру с точностью ±0,1 °С. Реакцию инициировали ферментом (10,1 мг/мл), предварительно разбавленным в 50 раз 0,1 М Трис-HCl буфером с рН 8,5. Для расчета удельной активности фермента использовали линейный участок кинетической кривой. При построении графика (рис. 6) использовали средние арифметические значения полученных удельных активностей.
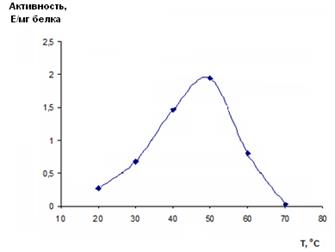
Рис. 6. Зависимость активности рекомбинантной имидазолонпропионат-аминогидролазы от температуры
Как следует из рисунка 6, температурный оптимум реакции наблюдается при температуре +50 °С. Активность фермента уменьшается более чем в два раза при +60 °С, причем такое снижение активности не может быть вызвано необратимой денатурацией фермента. Так, было показано, что при данной температуре фермент теряет 50% активности за 15 мин, тогда как время реакции в данной серии экспериментов составляет 4 минуты. Данный факт также подтверждается тем, что во время реакции не наблюдается значительных отклонений от линейности, как, очевидно, должно быть при денатурации белка в кювете.
Кинетические параметры рекомбинантного фермента, очищенного по разработанной схеме, рассчитанные на основании метода начальных скоростей, представлены в таблице 3 и на рисунке 7.
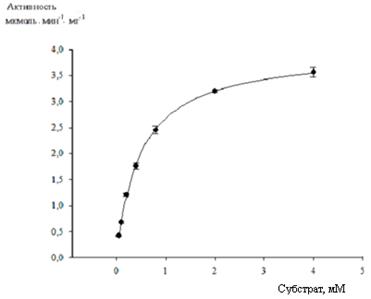
Рис. 7. Кривая насыщения субстратом имидазолонпропионат-аминогидролазы
Таблица 3
Зависимость активности имидазолонпропионат-аминогидролазы от концентрации субстрата в реакционной смеси
|
Концентрация субстрата, мМ |
Специфическая активность имидазолонпропионат-аминогидролазы, мкмоль×мин-1×мг-1 белка |
||
|
0,05 |
0,3961 |
0,4304 |
0,4266 |
|
0,10 |
0,6361 |
0,7161 |
0,6894 |
|
0,20 |
1,1732 |
1,2532 |
1,1922 |
|
0,40 |
1,7141 |
1,8664 |
1,6950 |
|
0,80 |
2,3312 |
2,5826 |
2,4416 |
|
2,00 |
3,1730 |
3,1692 |
3,2491 |
|
4,00 |
3,5896 |
3,7033 |
3,3911 |
Концентрация белка в реакционной смеси составляла 1,33 мкг/мл. Кинетические параметры имеют следующие значения:
Vmax (мкмоль мин-1 мг-1) = 3,9740 ± 0,056; Km (мM) = 0,4847 ± 0,021; kcat (с-1) = 5,193; kcat/Km (М-1 с-1) = 10713.
Заключение
1. Выделен hutI, кодирующий имидазолонпропионат-аминогидролазу, из клеточных экстрактов Pseudomonas putida B5437.
2. Клонирован ген hutI, кодирующий имидазолонпропионат-аминогидролазу.
3. Осуществлена экспрессия гена имидазолонпропионат-аминогидролазы в Escherichia coli BL21DE3.
4. Выделена и очищена рекомбинантная имидазолонпропионат-аминогидролаза Pseudomonas putida.
5. Изучены биохимические свойства рекомбинантной имидазолонпропионат-аминогидролазы Pseudomonas putida.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № Ор 15-38-50107/15 от 05.02.2015.
Рецензенты:
Короткий И.А., д.т.н., профессор, декан заочного факультета ФГБОУ ВО «Кемеровский технологический институт пищевой промышленности (университет)», г. Кемерово;
Бабич О.О., д.т.н., профессор кафедры «Бионанотехнология» ФГБОУ ВО «Кемеровский технологический институт пищевой промышленности (университет)», г. Кемерово.
Библиографическая ссылка
Каширских Е.В., Просеков А.Ю., Дышлюк Л.С., Носкова С.Ю., Карчин К.В., До Н.Д. ПОЛУЧЕНИЕ НОВОЙ АМИНОГИДРОЛАЗЫ PSEUDOMONAS PUTIDA И ХАРАКТЕРИСТИКА ЕЕ БИОХИМИЧЕСКИХ СВОЙСТВ // Современные проблемы науки и образования. 2015. № 5. ;URL: https://science-education.ru/ru/article/view?id=21442 (дата обращения: 29.06.2026).