



Уридинфосфорилаза является одним из ключевых ферментов метаболизма нуклеиновых кислот и удобной мишенью для химиотерапевтического воздействия. Уридинфосфорилаза многих возбудителей опасных инфекций отличается по субстратной специфичности от человеческой, в связи с чем ингибиторы уридинфосфорилазы рассматриваются как потенциальные лекарственные средства для лечения ряда инфекционных заболеваний. 5-Замещенные производные пиримидиновых нуклеозидов, например 1-(2-оксиэтоксиметил)-5-бензилурацил (бензилациклоуридин, BAU) и его аналоги, имеющие дополнительные заместители в ароматическом ядре, проявили выраженные ингибиторные свойства в отношении уридинфосфорилазы и усиливали противоопухолевое действие 2'-дезокси-5-фторуридина [10]. Описан синтез и ингибиторные свойства в отношении уридинфосфорилазы гетероцепных аналогов BAU - 1-(2-оксиэтоксиметил)производных 5-(фенилтио)- и 5-(фенилселено) урацила [9]. Однако активность соответствующих аза-аналогов - производных 5-(фениламино)урацила в отношении этого фермента ранее не была исследована.
Синтез разнообразных производных 5-(фениламино) урацила освещен в научной литературе достаточно широко. Известны производные 5-(фениламино) урацила, содержащие заместители бензильного [7] и арилоксиалкильного [8] типа, а также описаны некоторые ациклонуклеозиды на основе 5-(фениламино) урацила [5]. Некоторые из этих соединений проявляют заметную активность in vitro в отношении ВИЧ-1 [2-4] и вируса гепатита С [1]. Ингибиторные свойства этих соединений в отношении уридинфосфорилазы также ранее не изучались.
Цель исследования
Синтез и исследование ингибиторных свойств в отношении уридинфосфорилазы клеток печени человека ряда производных 5-(фениламино)ациклоуридина и их карбоцепных аналогов.
Материалы и методы исследования
Синтез 1-(2-оксиэтоксиметил)-5-(фениламино)урацила и его метилированных в ароматическое ядро аналогов был осуществлен по традиционной схеме синтеза «классических» ациклонуклеозидов, включающей региоселективное N1-алкилирование силилированного пиримидинового основания альфа-галоидэфиром - (2-бензоилоксиэтокси)хлорметаном с последующим удалением защитной бензоильной группы с помощью насыщенного при 0 °С метанольного раствора аммиака. Для алкилирования силилированного 5-(фениламино)урацила менее реакционноспособным 1-бром-4-ацетоксибутаном был использован метод «сплавления» путем нагревания реагентов без растворителя при температуре 180-185 °С в течение 1 ч, как это описано для других алкилирующих агентов с невысокой реакционной способностью [6].
1-(2-Бензоилоксиэтоксиметил)-5-(фениламино)урацил. Смесь 2,0 г (9,84 ммоль) 5-(фениламино)урацила, 50 мл гексаметилдисилазана, 1 г ацетамида и 0,5 г хлорида аммония кипятят с защитой от влаги воздуха в течение 8 ч до образования прозрачного раствора. Избыток гексаметилдисилазана упаривают в вакууме, остаток растворяют в 20 мл безводного 1,2-дихлорэтана, добавляют 2,2 г (10,2 ммоль) (2-бензоилоксиэтокси)хлорметана и перемешивают при комнатной температуре в течение суток. Последовательно добавляют 10 мл 2-пропанола, 20 мл воды и 5 мл концентрированного гидроксида аммония, перемешивают, органический слой отделяют, сушат сульфатом магния, фильтруют и упаривают в вакууме. Остаток перекристаллизовывают из смеси этилацетат - гексан (1 : 1) и получают 3,0 г светло-желтого кристаллического вещества, Т. пл. 95-98 °С, выход 80%. Спектр ЯМР 1Н, δ, м. д. (ДМСО-D6): 3,87 т (2 Н, 5 Гц, СН2); 4,36 т (2 Н, 5 Гц, СН2); 5,18 с (2 Н, N-CH2-O); 6,27 уш. с (1 Н, C5-NH); 6,55-7,18 м (5 Н, фенил); 7,30 с (1 Н, Н6); 7,18-8,00 м (5 Н, бензоил).
1-(2-Оксиэтоксиметил)-5-(фениламино)урацил (I). Смесь 1,5 г (3,93 ммоль) 1-(2-бензоилоксиэтоксиметил)-5-(фениламино)урацила и 25 мл насыщенного при 0 °С метанольного раствора аммиака выдерживают в герметично закрытом сосуде при комнатной температуре в течение суток, растворитель упаривают в вакууме, остаток промывают диэтиловым эфиром (2 ´ 15 мл), экстрагируют 50 мл кипящего 2-пропанола, горячий экстракт фильтруют, выдерживают при температуре -5 °С в течение суток, выделившийся белый кристаллический осадок отфильтровывают, промывают 10 мл диэтилового эфира и получают 0,8 г белого кристаллического вещества, Т. пл. 123-126 °С, выход 74%. Спектр ЯМР 1Н, δ, м. д. (ДМСО-D6): 3,52 с (4 Н, СН2СН2); 4,79 уш. с (1 Н, ОН); 5,10 с (2 Н, N-CH2-O); 6,60-7,22 м (6 Н, фенил, C5-NH); 7,46 с (1 Н, Н6).
Остальные соединения получают аналогично.
Активность полученных веществ в отношении уридинфосфорилазы клеток печени человека была изучена в Университете Алабамы (г. Бирмингем, Алабама, США) профессором Mahmoud H. el Kouni по методике [10].
Результаты исследования и их обсуждение
Синтез 5-(фениламино)ациклоуридина (I) и его гомологов (II-IV), содержащих дополнительные метильные группы в ароматическом ядре, а также соответствующих карбоцепных аналогов (V-VIII) был осуществлен по схеме (рис. 1).
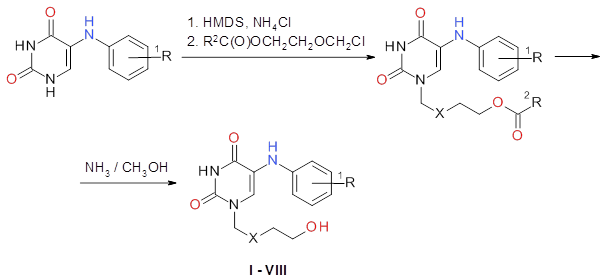
где: R1 = H, орто-, мета-, пара-CH3; R2 = CH3, C6H5; X = O, CH2
Рис. 1. Схема синтеза производных и аналогов 5-(фениламино)ациклоуридина.
Полученные соединения представляют собой белые кристаллические вещества, мало растворимые в воде, легко растворимые в спирте и ДМСО. Химическое строение соединений доказано методом ЯМР-спектроскопии. Выход и физико-химические свойства представлены в таблице 1.
Таблица 1
Выход и физико-химические свойства синтезированных соединений
|
Соединение |
R1 |
Х |
Брутто-формула |
Выход, % |
Т. пл., °С |
Rf * |
|
I |
Н |
O |
C13H15N3O4 |
74 |
123-126 |
0,56 |
|
II |
орто-CH3 |
O |
C14H17N3O4 |
76 |
165-167 |
0,56 |
|
III |
мета-CH3 |
O |
C14H17N3O4 |
68 |
122-124 |
0,53 |
|
IV |
пара-CH3 |
O |
C14H17N3O4 |
75 |
82-85 |
0,60 |
|
V |
Н |
CH2 |
C14H17N3O3 |
67 |
159-161 |
0,43 |
|
VI |
орто-CH3 |
CH2 |
C15H19N3O3 |
65 |
105-108 |
0,47 |
|
VII |
мета-CH3 |
CH2 |
C15H19N3O3 |
74 |
156-158 |
0,40 |
|
VIII |
пара-CH3 |
CH2 |
C15H19N3O3 |
79 |
146-150 |
0,42 |
* Примечание: Silufol UV-254, хлороформ - метанол (9 : 1).
Результаты исследования способности синтезированных соединений ингибировать уридинфосфорилазу клеток печени человека in vitro представлены в таблице 2.
Таблица 2
Ингибиторная активность синтезированных соединений в отношении уридинфосфорилазы in vitro
|
Соединение |
Ингибиторная концентрация, IC50, mМ |
|
I |
51,2 ± 4,2 |
|
II |
114,2 ± 9,5 |
|
III |
18,6 ± 1,0 |
|
IV |
112,7 ± 6,3 |
|
V |
83,1 ± 4,7 |
|
VI |
157,6 ± 17,7 |
|
VII |
18,3 ± 1,1 |
|
VIII |
131,2 ± 9,3 |
|
Бензилациклоуридин |
18,9 ± 1,4 |
Результаты исследований свидетельствуют о том, что незамещенный 5-(фениламино)ациклоуридин (I) и его карбоцепной аналог (V) соответственно в 2,7 и 4,3 раза уступают эталонному бензилациклоуридину по способности подавлять фосфорилазную активность фермента. Дополнительное введение в структуру этих веществ метильных групп в орто- и пара-положения еще больше уменьшают ингибиторную активность соединений II, IV, VI и VIII. Однако метилирование мета-положения дает обратный эффект: увеличение активности соединений III и VII по сравнению с их незамещенными аналогами I и V составляет 2,8 и 4,5 раза, и по величине ингибиторной концентрации IC50 1-(2-оксиэтоксиметил)-5-(мета-метилфениламино)урацил (III) и 1-(4-оксибутил)-5-(мета-метилфениламино)урацил (VII) не уступают бензилациклоуридину. Учитывая подобный характер влияния мета-замещения, можно предположить, что введение других заместителей (низших алкилов, арилов, галогенов) может значительно усилить ингибиторные свойства соединений, что свидетельствует о перспективности дальнейшего поиска новых высокоактивных в отношении уридинфосфорилазы веществ на основе 5-(фениламино)урацила.
Заключение
Осуществлен синтез аза-аналогов известного селективного ингибитора уридинфосфорилазы - бензилациклоуридина. Изменение природы линкера, связывающего ароматическое ядро и положение С5 ациклоуридина, с метиленовой группы на фрагмент вторичного амина приводит к снижению способности веществ угнетать фосфорилазную активность фермента, однако дополнительное введение метильной группы в мета-положение бензольного кольца полностью восстанавливает ингибиторные свойства как в случае с «классическим» производным ациклоуридина, так и в случае с карбоцепным аналогом. Представляет значительный теоретический и практический интерес синтез новых ациклических нуклеозидных аналогов на основе 5-(фениламино)урацила, имеющих разнообразные заместители в мета-положении, с целью поиска оригинальных высокоселективных ингибиторов уридинфосфорилазы.
Рецензенты:
Тюренков И.Н., д.м.н., профессор, заведующий кафедрой фармакологии и биофармации ФУВ ГБОУ ВПО «Волгоградский государственный медицинский университет» Минздрава России, г. Волгоград;
Сысуев Б.Б., д.фарм.н., доцент, заведующий научно-производственной лабораторией ГБУ «Волгоградский медицинский научный центр» Администрации Волгоградской области, г. Волгоград.
Библиографическая ссылка
Солодунова Г.Н., Маршалкин М.Ф., Соболевская С.И. СИНТЕЗ И ИНГИБИТОРНАЯ АКТИВНОСТЬ В ОТНОШЕНИИ УРИДИНФОСФОРИЛАЗЫ «КЛАССИЧЕСКИХ» И КАРБОЦЕПНЫХ АЦИКЛИЧЕСКИХ НУКЛЕОЗИДНЫХ АНАЛОГОВ – ПРОИЗВОДНЫХ 5-(ФЕНИЛАМИНО)УРАЦИЛА // Современные проблемы науки и образования. 2015. № 1-1. ;URL: https://science-education.ru/ru/article/view?id=17668 (дата обращения: 20.06.2026).