



Введение
Нейродегенерация – прогрессивная потеря структуры и функции нейронов, в конечном результате, приводящая к их гибели. Этот процесс является неотъемлемым этапом развития нервной системы и потому рассматривается как физиологически нормальный, однако в зрелом мозге при некоторых условиях он может переключиться в патологический. В таких случаях возникают нейродегенеративные болезни, которые составляют большую группу нейрологических заболеваний с гетерогенным клиническим и патологическим выражением. Несмотря на различия в клинической картине нейродегенеративных болезней, избирательной гибели отдельных популяций нейронов, некоторые механизмы на клеточном и молекулярном уровнях являются сходными. Известно, например, что при нейродегенеративных процессах различной природы наблюдаются признаки окислительного стресса, т. е. нарушения баланса между механизмами образования и элиминации активных форм кислорода, а также воспалительные явления. К общим признакам нейродегенеративных заболеваний относится длительный латентный период в их развитии [10]. Настоящий обзор посвящен эксайтотоксической гибели нейронов, возникающей в результате чрезмерной активации глутаматергической системы. Этот процесс наблюдается при многих нейродегенеративных заболеваниях [27,35,37], а также в условиях ишемии и травматических повреждений мозга [28].
Механизмы гибели нейронов
Согласно классификации «Комитета по клеточной гибели» (Nomenclature Committee on Cell Death) следует выделять типичные и нетипичные формы гибели клеток в организме [22]. Для мозга типичными формами гибели клеток являются апоптоз, некроз и аутофагия; нетипичными – некроптоз и эксайтотоксичность. Различие форм между собой определяется молекулярными механизмами и морфологическими особенностями клеточных изменений. Нейрональная гибель происходит в результате нормальных физиологических процессов в период развития, но также может быть индуцирована повреждающими факторами. Наиболее изучены механизмы апоптоза – программируемой гибели, которая регулируется эволюционно консервативными мессенджерами, состоящими из семейства каспаз, семейства Bcl-2 и белка Apaf-1 [4,40]. Выделяют два основных пути программированной гибели клеток: митохондриальный (присущей самой клетке) и через рецепторы гибели (внешний). Оба пути требуют вовлечения протеаз (каспаз), активность которых регулируется разными протеинами семейства Bcl-2. Показано, что клетки гибнут по такому пути при паркинсонизме и амиотрофическом латеральном склерозе (что выявлено при моделировании этих заболеваний на животных), предположительно также и при хорее Хантингтона [37]. Клетки, гибнущие в результате некроза, не следуют апоптотическому пути передачи сигнала, но при этом происходит активация различных рецепторов, что приводит к потере целостности клеточных мембран и неконтролируемому высвобождению продуктов клеточной гибели в межклеточное пространство. Это вызывает воспалительный ответ в окружающей ткани. Фактической причиной гибели при некрозе считают резкое падение содержания АТФ в клетках до такого уровня, который не совместим с жизнью [22]. К типу программированной гибели клеток относят аутофагию [22], которая является процессом лизосомальной деградации дефектных компонентов клетки и элиминирования поврежденных органелл и белковых агрегатов [29]. При аутофагии формируются специализированные структуры – аутофагосомы, внутри которых помещается клеточный материал (органелла или часть цитозоля), подлежащий разрушению. При слиянии аутофагосом с лизосомами образуются аутофаголизосомы, где происходит расщепление клеточного материала. Стимулами к запуску процессов аутофагии в клетках многоклеточных животных является отсутствие факторов роста, нехватка питательных веществ или наличие в цитоплазме поврежденных органелл, например, митохондрий, пероксисом и т. д.
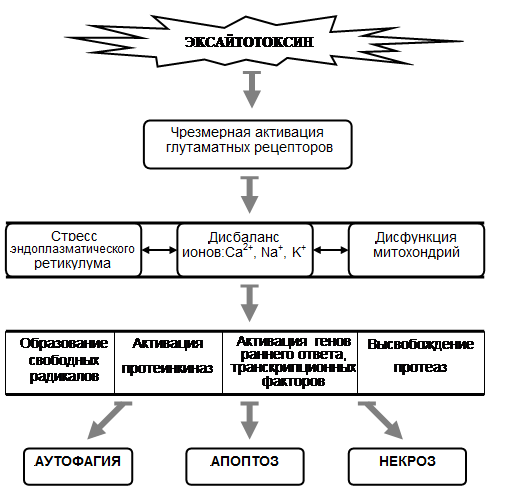
Рис. 1. Механизмы эксайтотоксичности
Эксайтотоксическую гибель нейронов относят к нетипичной форме гибели клеток [22], при которой наблюдается как апоптоз, так и некроз, а в некоторых условиях также аутофагия (рис. 1). Эксайтотоксичность возникает в результате чрезмерной или продолжительной активации рецепторов глутамата – основного возбуждающего нейромедиатора [27,38]. Гиперактивация ионотропных рецепторов глутамата приводит к входу Ca2+ в клетку; его высокая концентрация в цитоплазме запускает нейротоксические процессы, включающие разобщение митохондриальной транспортной цепи, активацию ряда ферментов, повреждающих нейроны [27,34]. Эксайтотоксичность является общим компонентом в механизмах ишемии [28] и многих нейродегенеративных заболеваний [27,35,38].
Экспериментальные исследования нейропротекции
Возможности самопроизвольного прекращения нейродегенеративных изменений мозговой ткани и полного восстановления нарушенных функций мозга, к сожалению, ограничены. Немногочисленные примеры показывают, что частичное восстановление возможно за счет ангиогенеза, компенсаторных процессов на клеточном и сетевом уровнях [28,31]. Например, кортикальная контузия у крыс через 48 часов приводит к частичной потере нейронов поля СА3 гиппокампа и снижению афферентов к нейронам СА1. В последующие недели происходит восстановление, хотя и не полное, афферентов к СА1 [30]. Несмотря на то, что свойства пластичности (развитие синаптической долговременной потенциации) в гиппокампе сохранялись, авторы наблюдали у животных дефекты памяти [30]. Функциональные нарушения, вызванные нейродегенеративными процессами, нормализовать удается не всегда, даже если гибель нейронов предотвращается. Примером может служить работа, в которой селективный ингибитор обратного захвата серотонина флуоксетин применяли у мышей после травматического повреждения мозга [39]. Авторы показали под влиянием флуоксетина улучшение нейропластических показателей и нейрогенеза в зубчатой фасции, однако ни локомоторная функция животных, ни способность их к пространственному обучению не восстановились.
Детальное изучение внутриклеточных механизмов при нейротоксичности и гибели клеток нервной ткани выявило ряд потенциальных мишеней, фармакологическое воздействие на которые представляется перспективным для поиска нейропротекторов. Одно из стратегических направлений поиска нейропротекторов основано на известных механизмах эксайтотоксичности. Ионотропные рецепторы глутамата привлекают особое внимание как мишени агентов, способных предотвратить эксайтотоксическое повреждение нейронов. Действительно, антагонисты NMDA-рецепторов показали нейропротективные свойства в условиях экспериментальной модели фокальной ишемии, однако, при глобальной ишемии они показали малую эффективность [9,23]. Кроме того, антагонисты NMDA рецепторов, например, МК-801 (Dizocilpine) вызывают каталепсию, моторные нарушения, ухудшают обучение [36]. Антагониcты АМРА рецепторов также обладают нежелательными побочными эффектами [9]. Поэтому клиническое применение блокаторов ионотропных рецепторов глутамата оказалось практически невозможным ввиду их малой эффективности или развития нежелательных побочных эффектов. Снижение эксайтотоксичности в экспериментальных условиях происходит при активации транспортеров глутамата, осуществляющих захват нейромедиатора из синаптической щели [8], однако клинического применения они также не нашли. Аналогичный вывод сделан и относительно блокаторов Са2+: фармакологическая блокада поступления Са2+ в нейроны в клинике нейродегенеративных заболеваний неэффективна [9, 34].
Важную роль в механизмах воспалительного ответа в мозге играет циклооксигеназа-2 (COX-2), уровень которой повышается при отсроченной нейрональной гибели. Ингибиторы COX-2 показали определенную эффективность в экспериментальной терапии повреждений мозга. Так, ингибитор DFU, введенный за 10 минут до травмы или в период менее чем 6 часов после нее, облегчает функциональное восстановление нейрологических рефлексов и памяти у крыс [17]. Нестероидный противовоспалительный препарат индометацин, также реализующий свои эффекты благодаря ингибированию СОХ-2, препятствует развитию у крыс функциональных нарушений, вызванных каиновой кислотой [2]. Мелатонин обладает нейропротективными свойствами в условиях эксайтотоксичности: его парентеральное введение после действия каиновой кислоты снижает внутриклеточное содержание Са2+ и оксида азота в нейронах, что способствует выживанию клеток [19]. Показано, что статины действуют как протекторы при некоторых условиях. Сравнительное исследование разных представителей статинов (simvastatin, lovastatin, fluvastatin, pravastatin, atorvastatin) в условиях эксайтотоксичности, вызванной каинатом у мышей, выявило наиболее эффективное нейропротекторное действие симвастатина, которое проявилось в снижении повреждения нейронов и апоптотической гибели клеток в гиппокампе и других структурах мозга. В этих экспериментах показано также позитивное влияние симвастатина на процессы памяти [32]. Стресс-белки обладают способностью увеличивать выживаемость клеток при разных повреждающих условиях. Экспериментальные исследования в моделях in vitro и in vivo показали нейропротективные свойства Hsp27 [3] и Hsp70 [16]. Повышенная экспрессия этих белков снижает гибель клеток как по типу апоптоза, так и некроза, уменьшает агрегацию протеинов в условиях ишемии. Индукция стресс-белков рассматривается как перспективный механизм в терапии болезней Альцгеймера и Хантингтона [21]. Нейропротекторными свойствами обладают также некоторые применяемые в клинике препараты, проявляющие активность одновременно в отношении нескольких клеточных мишеней. Особого внимания заслуживают противоэпилептические агенты, ввиду их способности снижать уровень возбуждения [24]. К таким препаратам относится, например, противоэпилептический агент вальпроат-натрий, который влияет на ГАМК-трансаминазу, потенциал-зависимые натриевые каналы, гликоген-синтазную киназу (GSK)-3, гистоновые деацетилазы [12]. Применение вальпроата после экспериментально вызванной травмы мозга показало его эффективность: у животных наблюдали улучшение состояния ГЭБ, уменьшение объема повреждения неокортекса, снижение гибели нейронов гиппокампа наряду с улучшением моторной функции и пространственной памяти животных. Постишемическое применение диазепама также снижает повреждение нейронов в поле СА1 гиппокампа [13], хотя происходит это за счет снижения температуры мозга. Клинические данные показывают также, что хроническое применение лития оказывает нейропротекторное действие [14]: у больных, терапия которых включает литий, увеличены объемы гиппокампа и амигдалы, а также снижен риск болезни Альцгеймера. Как уже подчеркивалось, ингибиторы ионотропных рецепторов нарушают когнитивные способности и вызывают нежелательные нейрологические симптомы [9]. Однако модуляция метаботропных рецепторов глутамата (mGlu) может быть перспективным направлением для нейропротекции [1,7]. Предлагается использовать лиганды разных подгрупп mGlu рецепторов в качестве фармакологических средств при некоторых заболеваниях: селективные агонисты mGlu3 рецепторов для терапии паркинсонизма, болезни Альцгеймера, амиотрофического латерального склероза; агонисты mGlu4 рецепторов для терапии двигательных расстройств [7]. Активация mGlu2/3 рецепторов производит тормозные эффекты, опосредованные снижением внутриклеточного цАМФ или снижением активности потенциал-зависимых Са2+-каналов [6], вследствие чего показаны нейропротекторные свойства их агонистов. Таким образом, на экспериментальных моделях многие фармакологические агенты проявляют нейропротекторные свойства, а в некоторых условиях наблюдается восстановление функций после повреждений. Однако их клиническое применение пока очень ограничено, что делает необходимым дальнейшее исследование механизмов нейроденерации и нейропротекции.
В 60-х годах прошлого века было показано, что обогащенная среда способствует функциональному восстановлению после повреждения мозга у экспериментальных животных. Последующие исследования детализировали и уточнили эти данные, прояснив некоторые молекулярно-клеточные механизмы. Изучение сравнительной эффективности компонентов среды показали, что когнитивная нагрузка (обучение) более эффективна, чем физические упражнения, а обогащение окружающей среды оказывает самое большое действие [41]. Механизмы, благодаря которым физические упражнения подавляют гибель нейронов, исследованы в работе [20]. Каиновая кислота была применена для индукции нейродегенеративных изменений в гиппокампе мышей. Усиленная тренировка животных на тредбане в течение 8 недель (1 час в день, 5 раз в неделю) снижала гибель нейронов, которая по предположению авторов обусловлена понижением уровня реактивных форм кислорода через активацию CREB, опосредуемую ERK1/2 и CaMKII. Обогащенная окружающая среда (физическая нагрузка, социальное взаимодействие и обучение) в раннем возрасте (начиная с 2–3 месяцев) оказала существенное протекторное действие при моделировании паркинсонизма на мышах [15]. Прекондиционирование различными факторами, при котором достигается состояние повышенной устойчивости к последующему повреждению (обусловленное активацией эндогенных адаптивных механизмов), также привлекает внимание многих исследователей. Показано, что применение сублетальных концентраций глутамата активирует через стимуляцию NMDA рецепторов эндогенные протективные механизмы мозга (NMDA-прекондиционирование) против глутаматной цитотоксичности и различных повреждающих воздействий [33]. Селективная фармакологическая активация этих механизмов рассматривается как перспективная терапия инсультов. Исследования на культуре нейронов показали, что нейропротекция с помощью NMDA-прекондиционирования связана с изменением экспрессии ряда белков, среди которых протеинкиназы, факторы транскрипции, ферменты антиоксидантной защиты.
Перспективным направлением в поиске нейропротекторов является изучение механизмов диеты с ограничением калорийности. Прогрессивная потеря нейронов, которая происходит при старении, снижается в результате применения такой диеты [18]. Необходимо отметить, что нейропротекторные свойства диеты зависят от экспериментальных условий, в частности от способа повреждения мозга. Так, гибель нейронов и функциональные нарушения снижались под влиянием диеты, если в эксперименте применяли каинат, но не другой нейротоксин – иботеновую кислоту [11]. Кроме того, диета не оказывает нейропротективного действия и не способствует восстановлению функций, если она применяется уже после повреждения мозга [26]. Известно, что диета с ограничением калорийности сопровождается повышением экспрессией регуляторного белка SIRT1 [42]. Регуляторные белки сиртуины (SIRTs или sirtuins) являются группой гистоновых деацетилаз. Нейропротективное действие сиртуинов было показано при острых и хронических нейрологических заболеваниях, таких как ишемия мозга, болезни Альцгеймера и Паркинсона, амиотрофического латерального склероза и множественного склероза. Механизмы действия сиртуинов не ограничиваются гистоновыми белками, они влияют также на ферменты репарации ДНК, протеинкиназы, транскрипционные факторы и коактиваторы [42].
Заключение
Явление эксайтотоксичности известно уже в течение нескольких десятилетий, однако молекулярно-клеточные механизмы прогрессивной гибели нейронов, происходящей при этом, недостаточно ясны, чтобы стать основой для создания эффективных средств для терапии нейрологических заболеваний. Успешно проявляющие свою эффективность в экспериментальных моделях нейропротекторы, такие как антагонисты NMDA и АМРА рецепторы глутамата, блокаторы кальциевых ионных каналов, блокаторы натриевых каналов, противовоспалительные средства, антиоксиданты пока не нашли широкого применения в клинике [9]. Поиск нейропротекторов, обладающих высокой эффективностью и минимальным побочным действием, активно продолжается. Одна из основных причин сложности создания нейропротекторов заключается в разветвленности путей дегенеративных процессов, влияющих друг на друга, и поэтому ингибирование одних путей не препятствует реализации других. Кроме того, показано, что нейродегенеративные процессы тесно связаны с иммунной системой организма [5,37], что усложняет представление об их механизмах и делает обоснованным положение о необходимости системных воздействий для успешной протекции. Молекулярно-клеточные процессы, участвующие в поддержании целостности нейрональных сетей при повреждении нервной ткани, включают ряд механизмов: синтез нейротрофических факторов и цитокинов; экспрессия белков, поддерживающих выживание клеток (шапероны, антиоксиданты, ингибиторы апоптоза и др.); активация белков, участвующих в репарации ДНК; мобилизация стволовых клеток для замены поврежденных нейронов и глии [25]. Для успешной терапии нейродегенеративных заболеваний важно найти способы активации эндогенных механизмов, препятствующих дегенеративным процессам, ингибирующих их развитие, если они индуцированы, восстанавливающих поврежденные функции.
Работа поддержана грантом РФФИ 12-04-00470.
Рецензенты:
Федотчев А. И., д.б.н., в.н.с. лаборатории механизмов рецепции Федерального государственного бюджетного учреждения науки Институт биофизики клетки РАН, г. Пущино.
Журавлева З. Н., д.б.н., в.н.с. лаборатории системной организации нейронов Федерального государственного бюджетного учреждения науки Институт теоретической и экспериментальной биофизики РАН, г. Пущино.
Библиографическая ссылка
Архипов В.И., Капралова М.В., Першина Е.В. ЭКСАЙТОТОКСИЧНОСТЬ И ЭКСПЕРИМЕНТАЛЬНЫЕ ПОДХОДЫ К НЕЙРОПРОТЕКЦИИ // Современные проблемы науки и образования. 2013. № 5. ;URL: https://science-education.ru/ru/article/view?id=10431 (дата обращения: 02.07.2026).