


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
QUANTUM CHEMICAL STUDY OF THE MECHANISM OF REACTION BETWEEN NITROGEN OXIDES WITH S – AND NITROSO – CONTAINING COMPOUNDS

Введение
Актуальной проблемой современной химической промышленности является разработка методов низкотемпературного окисления органических соединений, в том числе углеводородного сырья. Разработано большое число способов окисления. Среди них перспективными являются фотохимические методы. Так, например, известно, что ароматические нитросоединения могут выступать в качестве фотохимических окислителей, переносящих атом кислорода на окисляемую молекулу [3–9,10]. Интересным является и изучение самого механизма реакции переноса этого атома. Ароматические нитросоединения являются широко доступными промышленными продуктами. Кроме того, заманчива разработка методов окисления с их участием. Использование нитросоединений становится особенно актуальным, если принять во внимание то, что они часто являются основой взрывчатых веществ и их необходимо утилизировать.
Исследование фотохимического окисления серусодержащих соединений в присутствии нитросоединений в синглетном и триплетном состояниях при помощи методов компьютерного моделирования актуально. Работы, посвященные фотохимическим исследованиям реакций нитросоединений и нитрозооксидов с субстратами различной химической природы, были начаты нашей группой несколько лет назад [1,2].
Поиск переходного состояния
Поиск геометрического строения переходных состояний проведен при помощи процедур QST2 и QST3 из программного комплекса Gaussian03. Критерием корректности результатов расчета служило наличие только одной мнимой частоты для соответствующей молекулярной системы. Правильность нахождения пути реакции подтверждали при помощи успешного восстановления координаты реакции в рамках метода IRC. С помощью данного метода находятся два минимума энергии и один максимум, минимумы отвечают исходному и конечному состоянию, а максимум – переходному состоянию.
Переходные состояния реакций найдены при помощи методов TS, QST2 и QST3 из программного комплекса Gaussian-03w. Расчеты проводились, используя теорию функционала плотности (DFT) с полной оптимизацией геометрии гибридным методом Бекке–Ли–Янга–Парра (uB3LYP). Для осуществления расчетов использован программный комплекс Gaussian-03w.
Выбор метода расчета
Прежде чем производить квантово-химический расчет, необходимо выбрать метод и базис, с помощью которых он будет осуществлен. На данный момент существует огромное количество различных методов и базисов. Так, например, можно использовать методы молекулярной механики (MM+, AMBER, BIO), полуэмпирические методы MNDO, AM1, PM3, MP2, методы CASSCF. При выборе метода необходимо учитывать не только точность расчета, но и время, которое затрачивается на проведение этого расчета. При выборе метода нами использован принцип наилучшего соответствия геометрических параметров исследуемых объектов реакции с экспериментальными данными. Ниже приведены две таблицы с геометрическими параметрами нитрозосоединения (таблица 1) и серусодержащих соединений (таблица 2), соответственно. Из таблицы 1 видно, что среди перечисленных методов наиболее точно воспроизводит экспериментальные данные метод uB3LYP/6-31+G(d). Из таблицы 2 видно, что наилучшие результаты дал также метод uB3LYP/6-31+G(d). При выборе метода пришлось учитывать не только точность, но и время проведения расчета. Некоторые методы способны производить расчет с большей точностью, чем представленные выше, однако, для их реализации необходимо затратить больше машинного времени, чем для выбранного нами. Так, например, при использовании метода CASSCF время расчета может увеличиться от нескольких суток и даже месяцев.
Таблица 1. Выбор метода расчета для расчета нитрозосодержащих соединений (HNO)
|
Метод |
r (H-N), нм |
r (N-O), нм |
r (N-O) - r (H-N), нм |
|
uB3LYP/6-31+G(d)* |
0.1064 |
0.1208 |
0.0143 |
|
UHF/3-21G |
0.1036 |
0.1217 |
0.0181 |
|
UB3LYP/6-311+G(d) |
0.1064 |
0.1241 |
0.0177 |
|
PM3 |
0.0997 |
0.1175 |
0.0178 |
|
Эксперимент** |
0.1063 |
0.1211 |
0.0148 |
Таблица 2. Выбор метода расчета серусодержащих объектов
|
*Длины связей в нм |
MP2/6-31+G(d)*1 |
uB3LYP/6-31+G(d)*2 |
Эксперимент*3 |
|
SO(triplet) |
0.1524 |
0.1516 |
0.1481 |
|
SO2 |
0.1482 |
0.1466 |
0.1432 |
|
SO3 |
0.1463 |
0.1454 |
0.1430 |
|
SO2- |
0.1550 |
0.1548 |
0.1523 |
*1 Согласно нашей работе
*2 Nimlos, M. R.; Ellison, G. B. J. Phys. Chem. 1986, 90, 2574.[ACS Full Text ], [CAS]
*3 Chase, M. W., Jr.; Davies, C. A.; Downey, J. R., Jr.; Frurip, D. J.; McDonald, R. A.; Syverud, A. N. J. Phys. Chem. Ref. Data 1985, 14, Suppl. 1; JANAF Thermochemical Tables, 3rd ed.
Результаты и расчеты
В качестве объяснения полученных нами расчетных данных могут служить реакционные схемы (1) и (2).
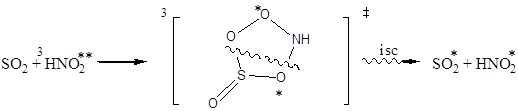 1
1
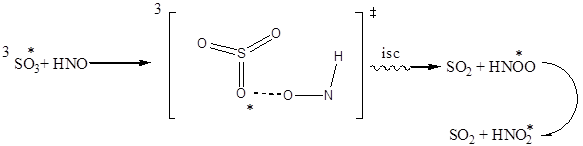 2
2
isc – интеркомбинационная конверсия.
Рассмотрим схемы исследуемых реакций.
Реакция 1 – это фотоокисление оксида серы (IV) молекулой нитросоединения в триплетном состоянии. В ходе реакции происходит отрыв атома кислорода и образование нитрозооксида, который затем превращается в нитросоединение. Реакция протекает, по-видимому, через пятичленное переходное состояние, причем выход продуктов реакции зависит от относительных величин энергий связи в переходном состоянии.
Реакция 2 является отрывом атома кислорода от триплетного триоксида серы и перенос его к молекуле нитрозосоединения. В ходе ее возможно также образование нитрозооксида с триплетном состоянии с последующей перегрпуппировкой в нитросоединение. Превращение нитрозооксида в нитросоединение было изучено авторами [7].
В таблице 4 приведены рисунки переходных состояний и энергетические характеристики соответствующих реакций в синглетном и триплетном состояниях.
Таблица 4. Переходные состояния рассматриваемых реакций
|
Синглетное переходное состояние, мнимая частота 226.83i см-1(S0)
|
Состояние молекулы |
Ea, ккал/моль |
Триплетное переходное состояние, мнимая частота 878.92i см-1(T1)
|
|
|
S0 |
115.22 |
|||
|
T1 |
66.25 |
|||
|
Синглетное переходное состояние, мнимая частота 537.25i см-1(S0)
|
Состояние молекулы |
Ea, ккал/моль |
Триплетное переходное состояние, мнимая частоты 461.92i см-1(T1)
|
|
|
S0 |
27.47 |
|||
|
T1 |
1.45 |
|||
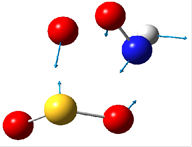
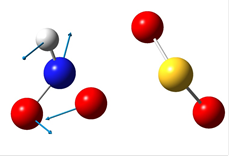
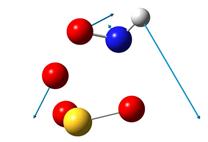
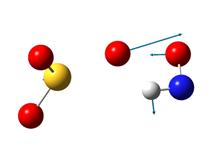
Стрелками указаны вектора смещений атомов. При рассмотрении направлений смещения атомов можно легко видеть, что атомы смещаются в сторону образования продуктов реакции. Кроме того, в таблице указаны частоты мнимых колебаний и энергии активации. Легко можно увидеть, что реакция протекает преимущественно в триплетном состоянии. Для реакции 1 энергия активации составляет 66.25 ккал/моль. Для реакции 2 – 1.45 ккал/моль.
Выводы
- На основании рассчитанных энергий активаций, реакции протекают преимущественно в триплетных состояниях с переносом атома кислорода.
- Реакции протекают через образование нитрозооксида преимущественно в триплетном состоянии.
- Рассмотрен один из возможных механизмов реакции фотоокисления нитросоединений в присутствии нитрозосоединений в разной мультиплетности.
- Правильность нахождения переходного состояния подтверждена наличием одной мнимой частоты.
Рецензенты:
Зеленцов Сергей Васильевич., д.х.н., профессор, зав. кафедрой фотохимии и спектроскопии ФГБОУ ВПО «Нижегородского государственного университета им. Н. И Лобачевского, 603950, г. Нижний Новгород.
Князев Александр Владимирович, д.х.н., профессор, Химический факультет ФГБОУ ВПО «Нижегородского государственного университета им. Н.И Лобачевского», г. Нижний Новгород.
Библиографическая ссылка
Плехович С.Д. КВАНТОВО-ХИМИЧЕСКОЕ ИЗУЧЕНИЕ МЕХАНИЗМОВ РЕАКЦИЙ ОКИСЛОВ АЗОТА С СЕРУСОДЕРЖАЩИМИ И НИТРОЗОСОДЕРЖАЩИМИ СОЕДИНЕНИЯМИ // Современные проблемы науки и образования. 2013. № 1. ;URL: https://science-education.ru/en/article/view?id=8365 (дата обращения: 04.08.2026).