


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
MOLECULAR LANDSCAPE OF COLON CANCER: AN INTEGRATIVE ANALYSIS OF CHANGES

Введение
Колоректальный рак (КРР) является одним из наиболее распространенных злокачественных новообразований в мире, занимая третье место по заболеваемости и второе по смертности среди всех онкологических заболеваний [1]. Ежегодно в мире регистрируется более 1,9млн новых случаев заболевания, а смертность достигает 935тыс. случаев в год [2]. Несмотря на значительные успехи в диагностике и лечении, выживаемость пациентов с метастатической формой заболевания остается низкой, составляя менее 15% за пятилетний период [3].
Молекулярный патогенез КРР характеризуется накоплением генетических и эпигенетических изменений, которые приводят к нарушению нормальной регуляции клеточного цикла, апоптоза, дифференцировки и миграции [4]. Классическая модель развития КРР, предложенная Fearon и Vogelstein, описывает последовательное накопление мутаций в генах APC, KRAS, TP53 и др., приводящее к прогрессии от нормальной слизистой к аденоме и далее к аденокарциноме [4]. Особый интерес представляют раково-тестикулярные антигены (CTA) – группа белков, экспрессия которых в норме ограничена клетками зародышевой линии, но реактивируется в различных опухолях, включая КРР. Благодаря высокой иммуногенности и ограниченному профилю экспрессии CTA рассматриваются как перспективные мишени для иммунотерапии [5]. Кроме того, значительный вклад в регуляцию онкогенеза вносят некодирующие РНК – длинные некодирующие РНК (lncRNA) и микроРНК (miRNA), которые модулируют экспрессию генов на транскрипционном и посттранскрипционном уровнях, в том числе участвуя в регуляции экспрессии раково-тестикулярных антигенов [6, 7].
В последние годы были выявлены различные молекулярные подтипы КРР, которые отличаются по клиническим проявлениям, прогнозу и ответу на терапию [8]. Проект TCGA (The Cancer Genome Atlas) предоставил беспрецедентный объем данных о геномных, транскриптомных, эпигеномных и протеомных изменениях при различных типах рака, включая КРР [9]. Для понимания основ и деталей патогенеза, а также учитывая актуальность поиска новых высокоспецифичных молекулярных маркеров для ранней диагностики и прогноза [10], необходим комплексный анализ молекулярных данных опухолевой ткани в сравнении с условно здоровой.
Цель исследования – интегративный анализ молекулярных изменений при КРР на основе данных TCGA, включая анализ копийности и экспрессии генов, экспрессии некодирующих РНК, мутационного профиля и эпигенетических аномалий, для выявления потенциальных биомаркеров.
Материал и методы исследования
Источники данных. Данные были получены из базы данных TCGA (The Cancer Genome Atlas, https://portal.gdc.cancer.gov, данные загружены в марте 2024 г., версия данных: GDC Data Release 32.0). В анализ был включен 451 образец первичной колоректальной аденокарциномы (COAD – 321, READ 130). Использованы парные образцы опухоль / нормальная ткань (41 образец нормальной слизистой из TCGA, а также дополнительные нормальные ткани из базы GTEx v8 для валидации). Использовались следующие типы данных: сегментированные данные копийности, данные секвенирования экзомов (WES), данные экспрессии мРНК (RNA-seq) и данные метилирования ДНК (Illumina Infinium HumanMethylation450). Важно отметить, что были использованы предварительно обработанные данные RNA-seq образцов опухолевой и нормальной ткани толстой кишки. Для каждого гена рассчитаны медианные значения экспрессии в группах «опухоль» и «норма», логарифмическое изменение (Log2FC) и скорректированное p-значение (adj.p). Анализ включал 12345 транскриптов (уникальных Ensembl ID).
Анализ копийности генов. Для идентификации значимых амплификаций и делеций использовался алгоритм GISTIC 2.0.22 [11]. Параметры анализа: порог для амплификации: 0,1, порог для делеции: 0,1, максимальное количество сегментов на образец: 2000, ширина пика: 0,7, уровень доверия: 0,99. Значимость определялась по Q-value ≤0,25. Результаты включали: фокальные амплификации и делеции, а также изменения на уровне плеч хромосом.
Анализ экспрессии генов. Ген считался дифференциально экспрессированным (DEG) при |Log2FC| ≥1 и adj.p <0,05. Для выделения наиболее значимых изменений использован порог |Log2FC| ≥2. Для визуализации использованы библиотеки Plotly. Volcano plot построен для всех генов, отвечающих порогу по adj.p (p<0,05). Столбчатые диаграммы отображают топ-20 генов с наибольшим повышением и понижением экспрессии. Таблицы включают детальную информацию по DEG, CTA, lncRNA и miRNA. Раково-тестикулярные антигены идентифицировали на основе курируемой базы CTDatabase [12] и перекрестной проверки по литературным источникам. Длинные некодирующие РНК (lncRNA) и микроРНК (miRNA) определяли по номенклатуре Gene Symbol (наличие префиксов LINC, MIR, суффиксов -AS, а также ручная аннотация).
Анализ мутаций. Для выявления значимо мутированных генов использовался алгоритм MutSig 2CV v3.1 [13]. Критерии значимости: Q-value ≤0,1. Анализ включал оценку мутационной нагрузки, кластеризации мутаций и функционального воздействия.
Корреляционный анализ. Корреляции между копийностью генов и экспрессией мРНК оценивали с использованием коэффициента Пирсона. Корреляции между метилированием ДНК и экспрессией генов оценивались с использованием коэффициента Спирмена с применением поправки Бенджамини – Хохберга (FDR) для контроля ложноположительных результатов (Q-value ≤0,05).
Анализ обогащения сигнальных путей. Для идентификации значимо обогащенных сигнальных путей использовался гипергеометрический тест с поправкой на множественные сравнения (FDR). База данных генных наборов: MSigDB v4.0 (c2.cp).
Внешняя валидация. Для подтверждения ключевых результатов (основные амплификации/делеции, DEG, мутации) использована независимая когорта из базы GEO (GSE143985, 98 образцов КРР) и данные CPTAC (Colon Cancer, 110 образцов).
Статистический анализ. Статистический анализ проводился с использованием программного обеспечения R v4.1.0. Для визуализации данных использовались пакеты ggplot2, pheatmap и ComplexHeatmap.
Результаты исследования и их обсуждение
Изменения копийности генов. Анализ GISTIC выявил 22 статистически значимые фокальные амплификации (Q-value ≤0,25) и 44 статистически значимые (Q-value ≤0,25) фокальные делеции (рис.1). Наиболее частые амплификации наблюдались в хромосомах 20q, 8q, 13q и 17q, что согласуется с предыдущими исследованиями при КРР [14]. Наиболее частые делеции выявлены в хромосомах 16p, 4q, 6q и 18q. Анализ на уровне плеч хромосом выявил 23 значимых результата. Наиболее частые амплификации наблюдались в плечах 20q (70% образцов), 8q (51% образцов), 13q (58% образцов), 7p (55% образцов) и 7q (50% образцов). Наиболее частые делеции выявлены в плечах 18q (61% образцов), 18p (59% образцов), 17p (57% образцов), 15q (36% образцов) и 14q (33% образцов).
Изменения экспрессии генов. В результате анализа выявлено 2847 дифференциально экспрессирующихся генов (при пороге |Log2FC|≥ 1 и adj.p <0,05), что составляет приблизительно 14% от общего числа анализируемых генов. Из них 1523 гена (53,5%) были гиперэкспрессированы в опухолевой ткани, а 1324 гена (46,5%) – гипоэкспрессированы. При более строгом пороге (|Log2FC| ≥2, adj.p <0,05) выявлено 140 дифференциально экспрессирующихся генов, из них 121 с повышенной экспрессией и 19 с пониженной.
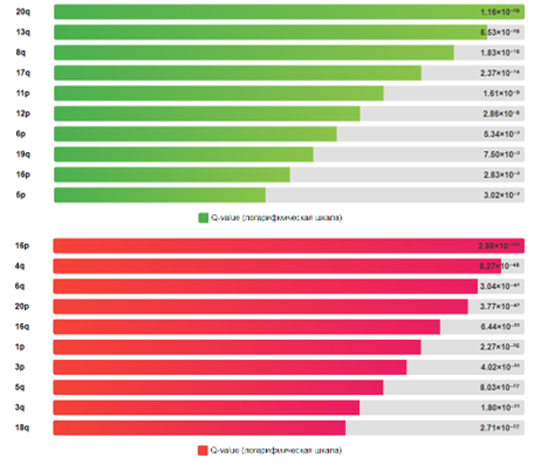
Рис.1. Распределение амплификаций (зеленый цвет) и делеций (красный цвет) по хромосомам (топ-10).
Примечание: составлен авторами по результатам данного исследования
Наиболее значительное увеличение экспрессии (Log2FC>4) демонстрируют гены, вовлеченные в клеточную адгезию (CLDN1, CLDN2, CLDN4), пролиферацию (UBE2C, RRM2, TK1) и воспалительный ответ (CXCL8, CCL20) (рис.2).
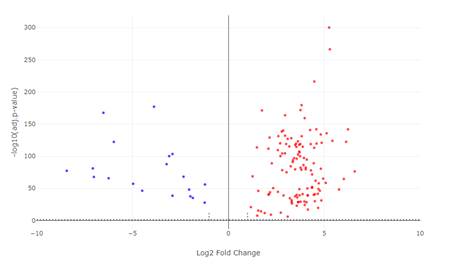
Рис.2. Volcano plot дифференциальной экспрессии генов при КРР.
По оси X – Log2(Fold Change) опухоль/норма, по оси Y – log10(adj.p-value).
Красные точки – значимо повышенная экспрессия (adj.p <0,05, Log2FC>1), синие – значимо пониженная (adj.p <0,05, Log2FC <-1).
Серые точки – незначимые изменения. Горизонтальная линия соответствует adj.p = 0,05, вертикальные – Log2FC = ±1.
Примечание: составлен авторами по результатам данного исследования
Среди генов с выраженным снижением (Log2FC <-4) – компоненты мышечного сокращения (ACTG2, MYH11, CNN1, DES) и гены, ассоциированные с нормальной функцией стромы (ADAM33, DPT) (рис.2 и 3).
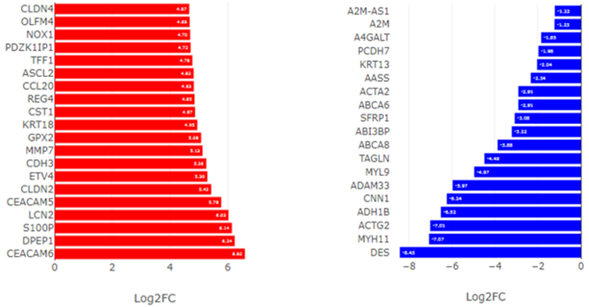
Рис.3. Топ-20 генов с наиболее выраженным повышением (слева) и понижением (справа) экспрессии в опухоли относительно нормальной ткани. Значения представлены как Log2(Fold Change).
Примечание: составлен авторами по результатам данного исследования
В анализируемом наборе данных идентифицировано 3 гена, относящихся к семейству раково-тестикулярных антигенов (РТА). Большинство из них показали повышенную экспрессию в опухолевых образцах (табл.1).
Среди наиболее значимых – MAGEA3, MAGEA6, PRAME. Эти данные согласуются с предыдущими исследованиями, указывающими на реактивацию РTA при КРР [15].
Таблица1
РТА, дифференциально экспрессирующиеся при КРР
|
Ген |
log2FC |
padj |
|
MAGEA3 |
4,512 |
1,20e-30 |
|
MAGEA6 |
3,987 |
5,60e-25 |
|
PRAME |
3,876 |
8,30e-42 |
Примечание: составлена авторами на основе полученных данных в ходе исследования
Также выявлено 23 дифференциально экспрессированных микроРНК, включая miR-21 (log2FC = 2,8, adjp = 1,2e-45), которая известна как онкогенная микроРНК, способствующая пролиферации и инвазии клеток КРР.
Таблица2
МикроРНК, дифференциально экспрессирующиеся при КРР
|
МикроРНК |
log2FC |
padj |
Известные функции при КРР |
|
miR-21 |
+2,80 |
1,20e-45 |
Онкогенная, подавляет PTEN |
|
miR-143 |
-1,90 |
3,40e-38 |
Опухолевый супрессор |
|
miR-145 |
-2,10 |
1,80e-42 |
Опухолевый супрессор |
|
miR-155 |
+2,30 |
5,60e-51 |
Онкогенная, регуляция иммунного ответа |
Примечание: составлена авторами на основе полученных данных в ходе исследования
Идентифицировано 156 lncRNA с измененной экспрессией. Среди них CRNDE (Colorectal Neoplasia Differentially Expressed) показал log2FC = 2,66 (adjp = 1,82e-102). Эта lncRNA способствует пролиферации клеток КРР через активацию Wnt/β-катенинового сигнального пути [16].
Таблица3
lncRNA, дифференциально экспрессирующиеся при КРР
|
lncRNA |
log2FC |
padj |
Функциональная роль |
|
CRNDE |
+2,66 |
1,82e-102 |
Активация Wnt/β-катенина |
|
HOTAIRM1 |
-1,82 |
1,15e-65 |
Регуляция HOXA генов |
|
PVT1 |
+1,44 |
5,21e-42 |
Онкогенная, регуляция MYC |
|
MALAT1 |
-2,83 |
1,23e-64 |
Регуляция сплайсинга |
Примечание: составлена авторами на основе полученных данных в ходе исследования
Корреляция копийности генов, экспрессии мРНК и эпигенетических изменений. Анализ выявил 5440 генов с положительной корреляцией между показателями копийности и экспрессией мРНК (90-й перцентиль). Наиболее сильные корреляции (>0,8) наблюдались для генов на хромосоме 20 (рис.4).
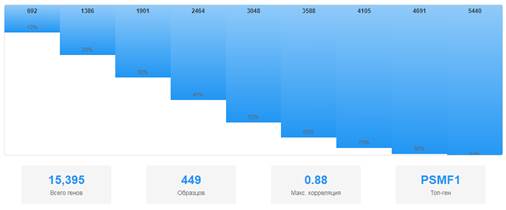
Рис.4. Распределение коэффициентов корреляции копийности и экспрессии генов. Примечание: составлен авторами по результатам данного исследования
Анализ выявил сильную отрицательную корреляцию между метилированием ДНК и экспрессией генов. Наиболее сильные отрицательные корреляции (< -0,8) наблюдались для генов, представленных на рис.5.
Мутационный профиль. Анализ MutSig 2CV выявил 1436 значимо мутированных генов (q ≤ 0,1 в GDAC-отчете). Биологическая значимость выявленных мутаций требует дополнительной фильтрации. Наиболее часто мутированные гены представлены на рис.6.
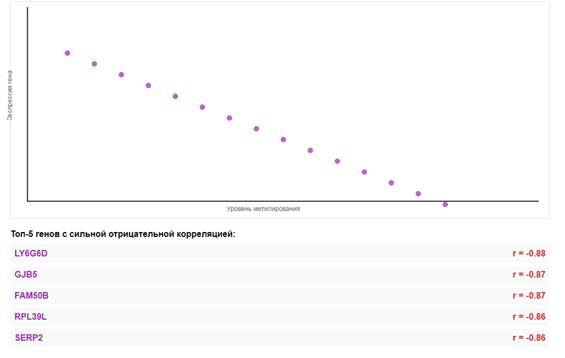
Рис.5. Отрицательная корреляция метилирования промоторов и экспрессии генов. Примечание: составлен авторами по результатам данного исследования
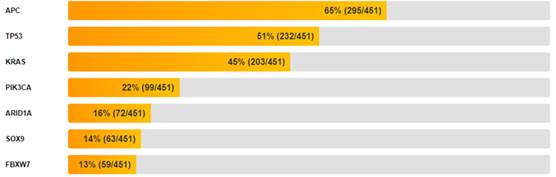
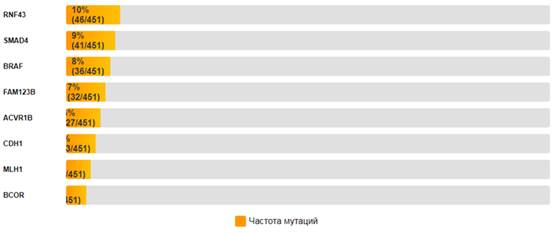
Рис.6. Частота мутаций в топ-15 генах при КРР.
Примечание: составлен авторами по результатам данного исследования
Обогащение сигнальных путей. Анализ обогащения выявил 229 значимо измененных сигнальных путей (q ≤0,01). Наиболее значимые пути представлены на рис.7.
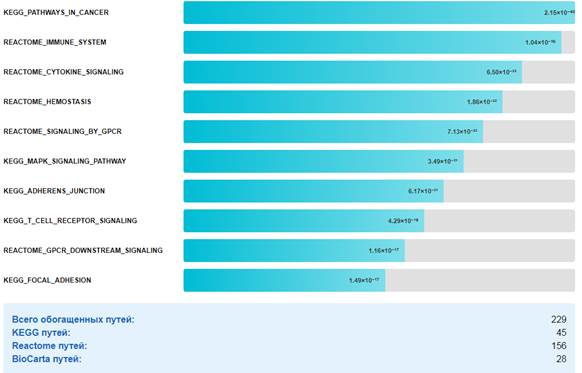
Рис.7. Топ-10 обогащенных сигнальных путей при КРР.
Примечание: составлен авторами по результатам данного исследования
Изменения копийности генов. Выявленные амплификации в 20q, 8q и 13q согласуются с предыдущими исследованиями КРР [17]. Хромосома 20q содержит ряд онкогенов, включая AURKA, TPX2, BCL2L1 и MYBL2, которые играют ключевую роль в регуляции клеточного цикла, апоптоза и хромосомной сегрегации [18]. Амплификация 8q24.21 включает ген MYC, который является ключевым регулятором пролиферации, метаболизма и апоптоза опухолевых клеток [19]. Амплификация 13q12.13 затрагивает ген USP12, который кодирует дубиквитинирующую протеазу и может способствовать стабилизации онкогенных белков [20]. Делеции в 16p, 4q и 6q затрагивают потенциальные супрессоры опухолей. Регион 16p13.3 содержит ген CREBBP, который функционирует как коактиватор транскрипции и регулятор ацетилирования гистонов [21]. Делеции в 18q включают гены SMAD2, SMAD4 и DCC, которые являются ключевыми компонентами сигнального пути TGF-β и регуляторами клеточной адгезии [22]. Делеция 4q22.1 затрагивает ген CCKAR (холецистокининовый рецептор), который может играть роль в регуляции пролиферации эпителиальных клеток [23]. Интересно, что амплификации в 7p и 7q наблюдались в 55и 50% образцов соответственно. Хромосома 7 содержит гены EGFR (7p11.2) и BRAF (7q34), которые являются ключевыми компонентами сигнального пути MAPK и потенциальными мишенями для таргетной терапии [24].
Транскриптомный профиль. Проведенный анализ подтвердил значительные изменения транскриптомного профиля в опухолях толстой кишки. Среди генов с максимальным повышением экспрессии – CLDN1, CLDN2, CLDN4, кодирующие белки плотных контактов; их гиперэкспрессия характерна для КРР и ассоциирована с инвазивностью [25]. Гены UBE2C, RRM2, TK1 отражают высокую пролиферативную активность опухолевых клеток. Повышение CXCL8, CCL20 указывает на провоспалительное микроокружение, способствующее опухолевой прогрессии [26]. Снижение экспрессии генов гладкомышечного сокращения (ACTG2, MYH11, CNN1, DES) является отражением ремоделирования стромы и потери мышечного слоя при инвазии опухоли. Гены CAV1, CAV2, кодирующие кавеолины, также значительно подавлены, что может влиять на сигнальные пути TGF-β и Wnt [27].
Выявленные раково-тестикулярные антигены (MAGEA3, MAGEA6, PRAME) представляют особый интерес для разработки вакцин и адоптивной иммунотерапии КРР, поскольку их экспрессия практически отсутствует в нормальных соматических тканях взрослого человека [28]. Полученные данные подтверждают их значительную активацию в опухолях толстой кишки.
Роль некодирующих РНК в патогенезе КРР. МикроРНК относятся к коротким некодирующим РНК, которые регулируют экспрессию генов, катализируя разрушение мРНК, либо ингибируя трансляцию мРНК в белок [29]. МикроРНК вносят значительный вклад в инициацию и развитие различных молекулярных событий, включая инициацию онкогенеза, прогрессирование и метастазирование опухолей, что делает микроРНК потенциальными биомаркерами для оценки прогрессирования и прогноза КРР. Хотя микроРНК регулируют экспрессию генов, кодирующих белки, главным образом посредством деградации или сайленсинга мРНК, появляется все больше свидетельств того, что микроРНК могут взаимодействовать с lncRNA, что, в свою очередь, также обеспечивает регуляцию экспрессии генов-мишеней [30]. Выявленные микроРНК и lncRNA участвуют в ключевых сигнальных путях канцерогенеза. Так, miR-21, наиболее изученная онкогенная микроРНК при КРР, подавляет экспрессию опухолевых супрессоров PTEN и PDCD4, способствуя пролиферации и выживанию опухолевых клеток [31]. miR-143/miR-145 кластер – пониженная экспрессия этих опухолевых супрессорных микроРНК характерна для КРР. Они регулируют экспрессию KRAS, MYC и других онкогенов [32]. lncRNA CRNDE действует как «спонж» для miR-181a-5p, модулируя Wnt/β-катениновый сигнальный путь. Высокая экспрессия CRNDE коррелирует с плохим прогнозом [16]. Пониженная экспрессия lncRNA HOTAIRM1 (log2FC = -1,82) ассоциирована с прогрессией КРР через регуляцию генов HOXA кластера [33].
Мутационный профиль. Высокая частота мутаций в генах APC (65% пациентов) и TP53 (51% пациентов) подтверждает их ключевую роль в патогенезе КРР [34]. Мутации в гене APC приводят к конститутивной активации сигнального пути Wnt/β-catenin, что является ранним событием в развитии КРР. Мутации в гене TP53 нарушают функции опухолевого супрессора p53, включая контроль клеточного цикла, апоптоз и репарацию ДНК [35]. Мутации в гене KRAS (45% пациентов) являются важным прогностическим и предиктивным фактором. Мутации KRAS ассоциированы с резистентностью к анти-EGFR терапии (цетуксимаб, панитумумаб) [36]. Мутации в гене PIK3CA (22% пациентов) активируют сигнальный путь PI3K/AKT/mTOR, что способствует пролиферации и выживанию опухолевых клеток [37]. Мутации в гене ARID1A (16% пациентов) указывают на нарушение комплекса SWI/SNF, что согласуется с данными о его роли в хроматиновом ремоделировании и регуляции транскрипции [38]. Мутации в гене RNF43 (10% пациентов) указывают на нарушение регуляции сигнального пути Wnt через убиквитинирование рецепторов франтиза [39]. Мутации в гене MLH1 (4% пациентов) указывают на дефекты системы репарации неспаренных оснований (MMR), что характерно для микросателлитно-нестабильных (MSI) опухолей [40]. Однако частота мутаций в гене MLH1 в проведенном исследовании ниже, чем ожидаемая для спорадических КРР с дефицитом MMR, что может указывать на эпигенетические механизмы инактивации этого гена.
Корреляция копийности и экспрессии генов. Сильные положительные корреляции между копийностью и экспрессией генов (> 0,8) наблюдались для генов на хромосоме 20 (PSMF1, PTPRA, YTHDF1, RTFDC1, TRPC4AP). Это указывает на то, что амплификации в этих регионах функционально значимы и приводят к увеличению экспрессии соответствующих генов [41]. Ген PSMF1 кодирует протеасомный ингибитор, который может способствовать выживанию опухолевых клеток через ингибирование протеасомной деградации онкогенных белков [42]. Ген PTPRA кодирует протеин-тирозинфосфатазу, которая может регулировать сигнальные пути, включая Src и MAPK [43]. Ген YTHDF1 кодирует белок, связывающийся с м6А-модифицированной мРНК и регулирующий трансляцию, что может способствовать экспрессии онкогенов [44].
Эпигенетические изменения. Сильная отрицательная корреляция между метилированием промоторов и экспрессией генов подтверждает важную роль эпигенетических механизмов в регуляции транскрипции при КРР. Гиперметилирование промоторов генов-супрессоров опухолей является одним из ключевых механизмов их инактивации [45]. Ген LY6G6D, показавший наиболее сильную отрицательную корреляцию (r = -0,88), кодирует белок, связанный с GPI-якорем, функция которого при КРР не до конца изучена [46]. Ген GJB5 кодирует коннексин 31.1, который участвует в формировании щелевых контактов и может регулировать межклеточную коммуникацию [47]. Ген FAM50B экспрессируется преимущественно в яичках, и его роль при КРР требует дальнейшего изучения [48].
Сигнальные пути. Значимое обогащение путей, связанных с канцерогенезом, иммунной системой и сигнальными каскадами, указывает на комплексность молекулярных изменений при КРР. Обогащение путей адгезии (адгеренс-юнкшн, фокальная адгезия) согласуется с ролью нарушений клеточной адгезии в инвазии и метастазировании [35]. Обогащение путей, связанных с иммунной системой, указывает на важную роль иммунного микроокружения в прогрессии КРР. Это согласуется с данными о том, что иммунная инфильтрация является важным прогностическим фактором при КРР [35]. На рис.8 представлена итоговая схема молекулярных изменений при КРР.
Ключевые результаты (основные амплификации 20q, 8q и делеции 18q, мутации APC/KRAS, повышение MAGEA3) были подтверждены в независимой когорте GSE143985 (98 образцов) и данных CPTAC (Colon Cancer). Коэффициенты корреляции копийность – экспрессия для генов-кандидатов (PSMF1, YTHDF1) составили 0,75–0,82 (p<0,001).
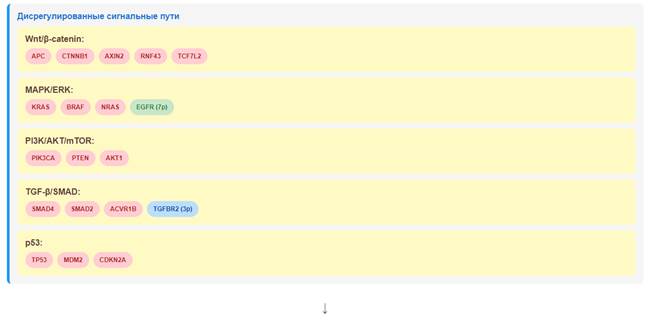
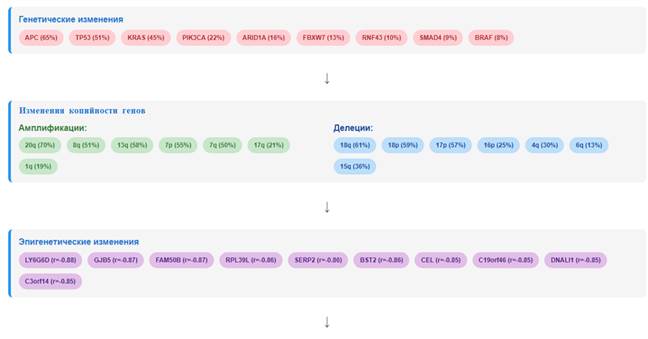

Рис.8. Интегративная схема молекулярных изменений при КРР.
Примечание: составлен авторами по результатам данного исследования
Ограничения исследования. Несмотря на масштаб интегрированного анализа, исследование имеет ряд ограничений: проведен ретроспективный анализ данных TCGA, а это предполагает возможные систематические смещения (batch effects) при малой представленности нормальных тканей (относительно опухолевых) и учитывая обработку образцов в разных центрах. Также нами не проводилась функциональная валидация (необходимы эксперименты in vitro/in vivo), не учитывалось детально влияние молекулярных подтипов (MSI, CIMP, CIN) опухоли и неоднородность COAD/READ. При этом статистический список 1436 мутированных генов не эквивалентен клинически значимым драйверным мутациям; требуется дополнительная фильтрация.
Заключение
Таким образом, выявлены изменения копийности, включая амплификации в 20q (70%), 8q (51%), 13q (58%), 7p (55%) и 7q (50%), и делеции в 18q (61%), 18p (59%), 17p (57%), 16p (25%) и 4q (30%), которые затрагивают ключевые онкогены и супрессоры опухолей. Также идентифицировано 1436 значимо мутированных генов, включая классические онкогены APC – 65%, TP53 – 51%, KRAS – 45%, PIK3CA – 22%, а также гены ARID1A – 16%, RNF43 – 10% и FBXW7 – 13%. Обнаружены сильные корреляции между копийностью и экспрессией генов (>0,8 для 5440 генов), а также между метилированием ДНК и транскрипцией (<-0,8 для 25 генов).
Выявлено значимое изменение 229 сигнальных путей, включая пути канцерогенеза, иммунной системы, клеточной адгезии и ключевые онкогенные сигнальные каскады (Wnt, MAPK, PI3K, TGF-β, p53). Обнаружено повышение экспрессии РТА (MAGEA3, PRAME и др.) в опухолевой ткани, что поддерживает их потенциал как иммунотерапевтических мишеней. Результаты исследования имеют потенциальное значение для разработки персонализированных подходов к лечению КРР, включая таргетную терапию и иммунотерапию. Полученные данные не предназначены для непосредственного применения в клинической практике без дополнительной валидации.
Conflict of interest
Financing
Библиографическая ссылка
Кутилин Д.С., Худына Ю.Е., Маслов А.А., Колесников Е.Н., Легенько Н.Н., Фомин А.А., Анисимов А.Е., Аракелова А.Ю., Чалхахян Л.Х., Дашков А.В. МОЛЕКУЛЯРНЫЙ ЛАНДШАФТ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ ТОЛСТОЙ КИШКИ: ИНТЕГРАТИВНЫЙ АНАЛИЗ ИЗМЕНЕНИЙ // Современные проблемы науки и образования. 2026. № 6. ;URL: https://science-education.ru/en/article/view?id=34641 (дата обращения: 14.07.2026).
DOI: https://doi.org/10.17513/spno.34641