


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
EVALUATION OF THE IMPACT OF NEK2 GENE KNOCKDOWN ON THE VIABILITY AND MIGRATION OF PROSTATE CANCER CELLS

Введение
Рак предстательной железы (РПЖ) остается одной из наиболее актуальных проблем онкологии. По данным мировой статистики за 2022 г., данное заболевание занимает второе место по частоте встречаемости среди всех злокачественных опухолей у мужчин и находится на пятом месте среди причин смертности от онкологических заболеваний в этой группе [1]. В Российской Федерации новообразования предстательной железы лидируют в структуре онкологической заболеваемости мужского населения, составляя 20,7% от общего числа случаев [2].
Несмотря на широкий арсенал современных методов лечения РПЖ, включающий радикальную простатэктомию, лучевую терапию, гормональную терапию (в том числе препаратами нового поколения, такими как абиратерон и энзалутамид), а также химиотерапию, основной проблемой, стоящей перед онкоурологией, остается развитие кастрационно-резистентной формы заболевания [3]. У 10–20% пациентов, страдающих РПЖ, в течение пяти лет развивается кастрационно-резистентный рак предстательной железы (КРРПЖ), что ассоциировано с неблагоприятным прогнозом, лекарственной устойчивостью и низкими показателями выживаемости [4]. Таким образом, поиск новых молекулярных мишеней и разработка инновационных терапевтических стратегий являются критически важными для повышения эффективности лечения этого заболевания.
Одной из таких перспективных мишеней в онкологических исследованиях выступает серин-треониновая киназа NEK2 (NIMA-related kinase 2), которая играет ключевую роль в регуляции митотического процесса [5]. Будучи гомологом митотического регулятора NIMA, обнаруженного у гриба Aspergillus nidulans, этот фермент локализуется в центросоме и участвует в контроле ее расщепления, формировании веретена деления, а также поддержании геномной стабильности в G2/M-фазах клеточного цикла [6, 7]. Первоначальные данные о повышенной экспрессии NEK2 в злокачественных опухолях были получены при изучении саркомы Юинга у детей [8]. Последующие исследования выявили ее сверхэкспрессию при различных онкологических заболеваниях, включая рак легких, молочной железы, колоректальный рак, рак простаты, желудка, печени и щитовидной железы [7]. Эта особенность тесно связана с хромосомной нестабильностью, возникновением анеуплоидии и развитием устойчивости опухолевых клеток к апоптозу. Избыточная активность NEK2 провоцирует преждевременное расщепление центросом, формирование аномальных веретен деления и способствует прогрессированию опухолевого процесса. Клинические данные указывают на прямую зависимость между уровнем экспрессии NEK2 и агрессивностью РПЖ, что подчеркивает ее потенциал как диагностического биомаркера и объекта для терапевтического воздействия [5]. Исследования рака желудка, поджелудочной железы и холангиокарциномы подтверждают онкогенную функцию NEK2, включая его участие в модуляции опухолевого микроокружения и процессах метастазирования [6]. Эксперименты на моделях рака яичников показали, что нокдаун NEK2 приводит к снижению миграционной активности клеток и блокирует активацию сигнального пути Wnt, индуцированную PCAT1. Эти результаты открывают новые возможности для разработки целевых методов терапии, направленных на нейтрализацию патологических механизмов, связанных с данной киназой [9].
Перспективным направлением для нейтрализации патологических механизмов, связанных со сверхэкспрессией NEK2, является применение технологии РНК-интерференции основанной на использовании малых интерферирующих РНК (миРНК). миРНК, представляющие собой двуцепочечные молекулы длиной 21–23 нуклеотида, позволяют с высокой специфичностью и эффективностью подавлять экспрессию целевых генов через деградацию комплементарной матричной РНК. Несмотря на существующие ограничения, такие как риск нецелевых эффектов, методика миРНК зарекомендовала себя как мощный инструмент функциональной геномики и перспективная основа для разработки терапевтических стратегий [10].
Цель исследования – оценить влияние нокдауна гена NEK2 с помощью миРНК на жизнеспособность и миграционную способность клеток РПЖ.
Материал и методы исследования
Для проведения исследования использовались две иммортализованные клеточные линии РПЖ: PC-3 и DU145, а также первичная культура клеток РПЖ, полученная от пациента после радикальной простатэктомии и подтвержденная патоморфологически.
Клеточные линии РПЖ человека (PC-3 и DU145) культивировали в модифицированной по способу Дульбекко среде Игла (DMEM; Servicebio, Китай) с добавлением 10% эмбриональной бычьей сыворотки (FBS; Himedia, Индия), содержащей 100 МЕ/мл пенициллина и 100 мкг/мл стрептомицина (ПанЭко, Россия), во влажной атмосфере при 37 °C в инкубаторе 8000 W (Thermo Fisher Scientific, США) с 5% CO2. Все клеточные линии прошли верификацию подлинности посредством STR-анализа с применением коммерческого набора COrDIS Plus (Гордиз, Россия) и тестирование на отсутствие микоплазменной контаминации методом ПЦР с использованием реагентов компании Servicebio (Китай).
Первичная культура РПЖ была получена из фрагмента опухоли, удаленной в ходе оперативного вмешательства, проводимого в качестве первого этапа по лечению РПЖ. Больной РПЖ проходил лечение в отделении онкоурологии ФГБУ «Национальный медицинский исследовательский центр онкологии» Министерства здравоохранения Российской Федерации в 2025 г. по поводу низкодифференцированной ацинарной аденокарциномы предстательной железы (Gleason 7: 4+3). Гистологический диагноз подтверждали в патологоанатомическом отделении ФГБУ «Национальный медицинский исследовательский центр онкологии» Министерства здравоохранения Российской Федерации. Пациент был осведомлен об участии в научном исследовании и подписывал информированное согласие на сбор биологического материала. Патологоанатомом в течение 10 мин после извлечения препарата выделялся фрагмент 500 мм3, соответствующий злокачественному компоненту опухоли, и помещался в раствор Хэнкса (Servicebio, Китай), содержащий 1% гентамицина (ПанЭко, Россия). Далее образец фрагментировали скальпелем до размера 1–2 мм3, после чего добавляли 300 ед/мл коллагеназы I (ПанЭко, Россия) в среде DMEM и инкубировали в течение 1ч при температуре 37 °C при постоянном перемешивании. По окончании ферментации образец дополнительно измельчали пипетированием и пропускали через стерильный нейлоновый фильтр (d=80 мкм) (Corning, США). Полученную суспензию дважды отмывали в фосфатном буфере и пассировали на культуральный флакон в среде DMEM, содержащей 10% FBS и 1% гентамицина и культивировали в стандартных условиях при 37 °C и 5% СО2.
Тотальную РНК выделяли из клеток РПЖ с помощью реагента TRIzol (Invitrogen, США) согласно протоколу производителя. Для повышения чистоты препаратов РНК проводили дополнительную очистку с применением набора RNeasy miniKit (Qiagen, Германия). Остаточную геномную ДНК удаляли инкубацией РНК с ДНК-азой I (Синтол, Россия). Концентрацию и чистоту выделенной РНК определяли количественно по ее поглощению при длине волны 260 нм с помощью флуориметра Qubit (Thermo Fisher, США). Синтез кДНК проводили методом обратной транскрипции с использованием набора MMLV Reverta (Синтол, Россия) и матричной РНК в качестве исходного материала. ОТ-ПЦР в режиме реального времени (ОТ-кПЦР) проводили с использованием реагента SYBR Green I (Синтол, Россия) согласно протоколу производителя, на приборе CFX 96 (Bio-Rad Laboratories, США). Последовательности праймерных пар, использованных для амплификации исследуемых генов, приведены в таблице, где представлена полная информация о нуклеотидных последовательностях. Последовательности специфических праймеров для NEK2 и GAPDH были основаны на соответствующей последовательности GenBank (NCBI) (GAPDH GeneID, NM_001256799.3; NEK2 GeneID, NM_002497.4).
Последовательности праймеров, используемых в ОТ-кПЦР
|
Ген |
Последовательность прямого праймера |
Последовательность обратного праймера |
|
GAPDH |
GATCATCAGCAATGCCTCCTG |
GAGTCCTTCCACGATACCAAAG |
|
NEK2 |
CCACAGACGAAGTGATGGTG |
TGATTTTCCCAGCGAGTTCT |
Примечание: составлена авторами на основе полученных данных в ходе исследования
ПЦР проводили при следующих условиях: начальная денатурация при 95 °C в течение 5 мин, затем 35 циклов по 95 °C в течение 30 с, 55 °C в течение 1 мин и 72 °C в течение 1 мин, с заключительным этапом удлинения при 72 °C в течение 5 мин. Специфичность амплификации подтверждали анализом кривых плавления (melt curve): во всех реакциях регистрировали один пик, что свидетельствовало об отсутствии неспецифичных продуктов и праймер-димеров. В каждую постановку включали отрицательный контроль без матрицы (NTC), в котором амплификация отсутствовала либо превышала рабочий диапазон не менее чем на 3 цикла. Уровни экспрессии мРНК NEK2 нормализовали по уровням GAPDH. Эффективность амплификации праймеров находилась в допустимых пределах (90–110%) и была сопоставима для таргетного гена NEK2 и референсного гена GAPDH. Для оценки экспрессии применялся показатель ΔCt. Все эксперименты ПЦР повторяли 3 раза.
Нокдаун гена NEK2 осуществляли с использованием синтетических миРНК, сконструированных для специфического подавления экспрессии транскрипта NEK2 (RefSeq: NM_002497.4). Последовательности направленных миРНК включали смысловую цепь (5′-UAGUAACGAACGAUGUUUGGA-3′) и антисмысловую цепь (5′-CAAACAUCGUUCGUUACUAUG-3′). Дизайн миРНК выполняли с применением онлайн-платформы siDirect v2.1 с соблюдением алгоритма Ui-Tei для минимизации нецелевых эффектов [11–13]. В качестве отрицательного контроля использовали неспецифическую миРНК (QIAGEN, США).
Трансфекцию проводили с помощью реагента Lipofectamine RNAiMAX (Invitrogen, США) согласно стандартному протоколу производителя. За 24ч до трансфекции клетки РПЖ высевали в 6-луночные планшеты из расчета 0,5×106 клеток на лунку, чтобы достичь 60–80% конфлюэнтности к моменту трансфекции.
Для приготовления трансфекционных комплексов использовали бессывороточную среду Opti-MEM (Gibco, США). Сначала готовили два отдельных раствора: раствор А – 10, 25, 50 или 100 нМ миРНК в 150 мкл Opti-MEM, раствор Б – 7,5 мкл Lipofectamine RNAiMAX в 150 мкл Opti-MEM. Оба раствора инкубировали при комнатной температуре в течение 5 мин, после чего объединяли в соотношении 1:1 и дополнительно инкубировали 20 мин для формирования комплексов миРНК-Lipofectamine. Полученную смесь объемом 250 мкл на лунку добавляли непосредственно к клеткам, находящимся в полной культуральной среде. Клетки инкубировали в течение 6ч в стандартных условиях (37 °C, 5% CO2), после чего среду заменяли на свежую полную культуральную среду для продолжения инкубации.
Для определения оптимальных параметров подавления NEK2 оценивали эффективность нокдауна при различных концентрациях миРНК (10–100 нМ) и временных точках (24, 48 и 72ч после трансфекции). Анализ проводили методом ОТ-кПЦР с использованием специфичных праймеров к гену NEK2. Уровень подавления рассчитывали по формуле
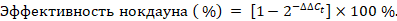
Максимальное снижение экспрессии NEK2 на уровне мРНК выбрано в качестве критерия для последующих функциональных экспериментов.
Жизнеспособность клеток после трансфекции оценивалась с использованием трипанового синего (Биолот, Россия) через 24, 48, 72 и 96ч. Клетки собирали, ресуспендировали в 0,4% растворе трипанового синего в соотношении 1:1 и подсчитывали количество живых (незакрашенных) и мертвых (синих) клеток в камере Горяева под микроскопом DM IL LED (Leica, Германия). Живые клетки выражались в процентах от общего числа клеток.
Для анализа влияния подавления экспрессии гена NEK2 на миграционные свойства клеток был выполнен скретч-тест (wound healing assay). Клетки высевали в 24-луночные культуральные планшеты с плотностью 1×105 клеток на лунку и культивировали в инкубаторе при 37 °С и 5 % CO2 в течение 24ч до достижения полного конфлюэнтного монослоя. После формирования сплошного слоя клеточный монослой повреждали путем проведения стерильным наконечником для пипетки объемом 100 мкл по центру лунки, создавая искусственную «рану». Затем свободную от клеток область аккуратно промывали фосфатно-солевым буфером (PBS) (Servicebio, Китай) для удаления отторгнутых клеток и обломков. В тот же момент проводили трансфекцию клеток миРНК, направленной против NEK2.
Визуализацию зоны повреждения осуществляли с помощью инвертированного микроскопа DM IL LED (Leica, Германия) в момент времени 0ч, а затем через 24, 48, 72 и 96ч после создания «раны». Площадь клеточно-свободной области измеряли с использованием программного обеспечения ImageJ (NIH, США). Скорость миграции оценивали по степени закрытия дефекта относительно начального размера «раны» в течение 96ч. Для каждой экспериментальной точки анализировали несколько изображений из разных участков одной лунки для обеспечения достоверности данных.
В качестве отрицательных контролей использовали немодифицированные клетки и клетки, трансфицированные контрольной миРНК, не специфичной к целевому гену.
Все эксперименты выполнялись в трехкратной биологической и технической повторности. Полученные данные представлены в виде среднего значения и стандартного отклонения (M (SD)). Статистическую обработку данных выполняли в среде R (версия 4.4.2). Нормальность распределения внутри каждой группы оценивали критерием Шапиро – Уилка, гомогенность дисперсий – тестом Левена. Для сравнения базального уровня экспрессии гена NEK2 между клеточными линиями применяли однофакторный дисперсионный анализ Уэлча (Welch ANOVA), так как было выявлено нарушение предпосылки о равенстве дисперсий. Для последующих попарных сравнений между группами использовали критерий Games – Howell, устойчивый к неоднородности дисперсий и различиям объемов выборок. Анализ эффективности нокдауна, жизнеспособности и миграционной активности клеток проводили с использованием двухфакторного дисперсионного анализа для данных, преобразованных методом выравненных рангов (Aligned Rank Transform, ART), реализованного в пакете ARTool. Для апостериорного анализа использовали метод наименьших средних квадратов (estimated marginal means, пакет emmeans) с последующим выполнением попарных сравнений. Коррекцию уровня значимости при множественных сравнениях осуществляли методом Холма. Статистически значимыми считали различия при уровне p<0,05.
Результаты исследования и их обсуждение
Сравнительный анализ базальной экспрессии гена NEK2 в клеточных линиях PC-3, DU145 и первичной культуре PCa-PC выявил статистически значимые различия между исследуемыми моделями (рис.1). Уровень экспрессии NEK2 в первичной культуре PCa-PC был достоверно выше по сравнению с клеточными линиями PC-3 и DU145 (p<0,001 для обоих сравнений). Кроме того, в линии DU145 экспрессия NEK2 была статистически значимо выше, чем в линии PC-3 (p<0,05).
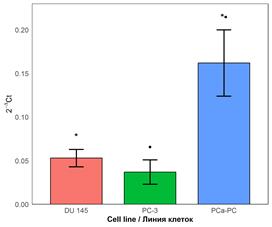
Рис.1. Относительный уровень экспрессии гена NEK2 в клеточных линиях РПЖ.
Примечание: * – статистически значимое различие по сравнению с линией PC-3 (p<0,05); • – статистически значимое различие по сравнению с линией DU145 (p<0,05).
Примечание: составлен авторами по результатам данного исследования
Наименьший уровень экспрессии NEK2 был зафиксирован в клеточной линии PC-3 и составил 0,037 (0,014). В клетках линии DU145 данный показатель был выше и достигал 0,053 (0,010). Наиболее высокий уровень экспрессии NEK2 наблюдался в первичной культуре PCa-PC и составлял 0,162 (0,038). Уровень экспрессии NEK2 в PCa-PC превышал соответствующие значения в линии PC-3 в 4,4 раза и в линии DU145 – в 3,1 раза, что подчеркивает выраженную активацию данного гена в первичной опухолевой культуре.
Эффективность подавления экспрессии гена NEK2 во всех исследованных моделях достоверно зависела от концентрации миРНК и времени после трансфекции (p<0,001), при отсутствии значимого взаимодействия между этими факторами (p>0,05), что указывает на их независимый вклад в формирование эффекта нокдауна.
В клеточной линии PC-3 концентрации 50 и 100 нМ обеспечивали значительно более выраженное подавление экспрессии NEK2 по сравнению с 10 нМ (p<0,01), тогда как различия между 50 и 100 нМ были статистически незначимыми (p>0,05). Максимальная эффективность наблюдалась через 48ч, при этом доза 50 нМ характеризовалась наибольшей стабильностью и воспроизводимостью эффекта, тогда как при 100 нМ, особенно к 72ч, возрастала внутригрупповая вариабельность.
В линии DU145 влияние концентрации миРНК было наиболее выраженным (p<2× 10-16), при сохранении значимого, но менее выраженного эффекта времени (p<0,001). Уже через 24ч дозы 50 и 100 нМ обеспечивали существенно более высокий нокдаун по сравнению с 10 и 25 нМ (p<0,001), а различия между 50 и 100 нМ во всех временных точках отсутствовали (p>0,05), что свидетельствует о насыщении эффекта при 50 нМ.
В первичной культуре PCa-PC также было выявлено значимое влияние концентрации (p < 10-14) и времени (p<0,001) при отсутствии их взаимодействия (p≈0,98). Дозы 50 и 100 нМ во всех временных точках вызывали более выраженное подавление экспрессии NEK2 по сравнению с 10 нМ (p<0,001) и, в большинстве случаев, с 25 нМ (p<0,01), при отсутствии различий между 50 и 100 нМ (p>0,05). Для этих концентраций отмечалось усиление эффекта к 72ч по сравнению с 24ч.
Низкие концентрации миРНК (10–25 нМ) во всех моделях вызывали слабый и нестабильный нокдаун экспрессии NEK2, что ограничивает их практическую применимость. Контрольная неспецифическая миРНК (NC-siRNA) не оказывала направленного влияния на уровень экспрессии NEK2, подтверждая специфичность наблюдаемого эффекта.
Таким образом, концентрация 50 нМ миРНК была выбрана для дальнейших экспериментов как оптимальная, поскольку она обеспечивала максимальный и статистически значимый нокдаун экспрессии NEK2, сопоставимый с эффектом 100 нМ, но при меньшей вариабельности и большей воспроизводимости результатов.
Анализ жизнеспособности клеток показал, что нокдаун гена NEK2 приводит к выраженному и статистически значимому снижению числа живых клеток во всех исследованных моделях по сравнению с контролем и NC-siRNA (p<0,001). Для всех линий выявлено достоверное влияние как фактора обработки, так и времени инкубации, а также их взаимодействия, что указывает на выраженную времязависимую модуляцию эффекта нокдауна (рис.2). В клеточной линии PC-3 уже через 24ч после трансфекции жизнеспособность клеток при обработке 50 нМ миРНК была существенно ниже по сравнению с контролем и NC-siRNA (p<0,001), и это различие сохранялось на всех последующих временных точках. Несмотря на рост клеточной популяции в обеих группах, скорость пролиферации при нокдауне NEK2 была значительно ниже. К 96ч количество жизнеспособных клеток в экспериментальной группе было примерно вдвое меньше, чем в контроле (p<0,001). Аналогичная картина наблюдалась в линии DU145. Различия между контролем, NC-siRNA и 50 нМ миРНК были статистически значимыми уже через 24ч и сохранялись на протяжении всего эксперимента (p<0,001). К 96ч жизнеспособность клеток при нокдауне NEK2 была снижена примерно вдвое по сравнению с контролем, что свидетельствует о выраженном ингибирующем влиянии подавления NEK2 на рост и выживание клеток данной линии. Наиболее выраженный эффект был зафиксирован в первичной культуре PCa-PC. Уже через 24ч после трансфекции наблюдалось значимое снижение жизнеспособности клеток в экспериментальной группе по сравнению с контролем (p<0,001), которое сохранялось и усиливалось в динамике. К 96ч количество живых клеток при обработке 50 нМ миРНК было снижено более чем в 2,4 раза относительно NC-siRNA и контроля (p<0,001), что указывает на высокую чувствительность первичной культуры к нокдауну NEK2. Во всех моделях контрольные культуры демонстрировали активный и статистически значимый рост клеточной популяции во времени, тогда как при подавлении NEK2 пролиферация сохранялась, но протекала существенно медленнее. Совокупно полученные данные подтверждают, что NEK2 играет ключевую роль в поддержании пролиферативной активности и жизнеспособности клеток РПЖ, а его подавление приводит к устойчивому и воспроизводимому снижению жизнеспособности как опухолевых линий, так и первичной культуры.
Скретч-тест показал, что нокдаун гена NEK2 вызывает выраженное и статистически значимое снижение миграционной активности клеток во всех исследованных моделях (p<0,001), при достоверном влиянии как фактора обработки, так и времени, а также их взаимодействия (рис.2).
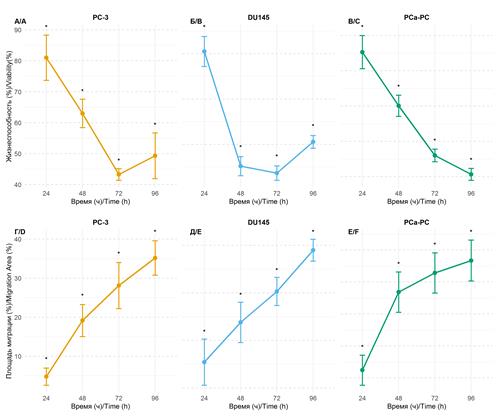
Рис.2. Динамика жизнеспособности и миграционной активности клеточных линий РПЖ во времени. А – PC-3 (жизнеспособность); Б – DU145 (жизнеспособность); В – PCa-PC (жизнеспособность); Г– PC-3 (миграция); Д – DU145 (миграция); Е – PCa-PC (миграция).
Примечание: составлен авторами на основе полученных данных в ходе исследования.
* статистически значимое различие по сравнению с контролем (p<0,001)
В линии PC-3 площадь закрытия раны снижалась с 40,22% в контроле до 4,80% при нокдауне через 24ч и с 95,28 до 35,18% к 96ч, что отражает более чем двукратное уменьшение миграционной активности. В линии DU145 аналогично через 24ч миграция составляла 10,91% против 39,98% в контроле, а к 96ч – 57,98% против 94,09%, что также соответствует почти двукратному снижению. В первичной культуре PCa-PC контрольные клетки достигали максимального закрытия раны 76,90% к 72ч и 70,73% к 96ч, тогда как при нокдауне NEK2 миграция оставалась значительно ниже и составляла 12,43% через 24ч и 46,71% к концу эксперимента. В совокупности полученные данные свидетельствуют о том, что подавление экспрессии NEK2 резко и устойчиво ингибирует миграционную способность клеток рака предстательной железы, проявляясь уже в первые сутки и сохраняясь на протяжении всего периода наблюдения.
Полученные данные подтверждают, что серин-треониновая киназа NEK2 играет важную роль в поддержании онкогенных свойств клеток РПЖ. Нокдаун гена NEK2 с использованием РНК-интерференции приводил к снижению жизнеспособности и миграционной активности клеток, причем эффект воспроизводимо наблюдался во всех трех моделях, включая первичную культуру, полученную от пациента с низкодифференцированной ацинарной аденокарциномой (Глисон 7: 4+3). Такая согласованность результатов в разных системах подчеркивает потенциальную универсальность NEK2 как терапевтической мишени [5–9].
Наиболее значимым наблюдением стало снижение жизнеспособности после подавления NEK2 во всех моделях, что согласуется с работами, где высокая экспрессия NEK2 необходима для пролиферации опухолей различных локализаций [5]. Вероятно, вклад NEK2 не ограничивается «классической» функцией в регуляции митоза: нарушение целостности центросом и сборки веретена деления может приводить к остановке клеточного цикла в фазе G2/M и запуску гибели клеток. Однако выраженность эффекта позволяет предполагать более сложную, многоуровневую регуляцию опухолевой выживаемости [6].
Одним из таких уровней может быть участие NEK2 в регуляции альтернативного сплайсинга. Согласно литературным данным, NEK2 способен фосфорилировать сплайсинг-фактор SRSF1, что смещает баланс в сторону образования антиапоптотических изоформ различных генов. Подавление экспрессии NEK2 приводит к увеличению уровня проапоптотических вариантов генов-мишеней SRSF1 и повышает чувствительность клеток к апоптозу. Кроме того, показано, что NEK2 участвует в метаболическом перепрограммировании опухолевых клеток за счет сплайсинг-переключения гена PKM с изоформы PKM1 на PKM2, что поддерживает гликолитический метаболизм, характерный для раковых клеток. Таким образом, наблюдаемое в исследовании снижение жизнеспособности клеток, вероятно, обусловлено одновременно митотическими нарушениями и дисрегуляцией альтернативного сплайсинга, затрагивающей как апоптотические, так и метаболические пути [14–15].
Снижение миграционной способности клеток, выявленное в скретч-тесте, было выраженным и сохранялось на протяжении 96ч, что свидетельствует о стабильности наблюдаемого эффекта. Полученные данные согласуются с имеющимися сведениями о роли NEK2 в активации канонического пути Wnt/β-катенин: NEK2 способствует снижению уровня E-кадгерина и индуцирует экспрессию мезенхимальных маркеров, таких как N-кадгерин, тем самым запуская эпителиально-мезенхимальный переход (EMT). Этот процесс ассоциирован с повышением инвазивности и метастатического потенциала опухолевых клеток. В различных экспериментальных моделях ингибирование NEK2 приводит к уменьшению ядерной фракции β-катенина и снижению экспрессии его транскрипционных мишеней – в частности, Cyclin D1 и c-Myc, – а также к восстановлению функции деструктивного комплекса β-катенина (включающего GSK-3β и APC). В совокупности это сопровождается подавлением EMT и восстановлением эпителиального фенотипа [16].
Еще одним важным механизмом действия NEK2 является модуляция оси E2F1–IGF2 в плоскоклеточном раке пищевода (ESCC). NEK2 конкурирует с CDC20 за связывание с транскрипционным фактором E2F1, тем самым защищая его от деградации. При подавлении NEK2 деградация E2F1 усиливается, что приводит к снижению транскрипции IGF2 и замедлению пролиферации опухолевых клеток [17].
Ряд дополнительных исследований указывает на участие NEK2 и в других онкогенных сигнальных путях, таких как ERK/MAPK, AKT и Hippo, а также на ее способность усиливать прометастатические программы альтернативного сплайсинга – например, посредством взаимодействия с белком Sam68 [6, 16, 18]. Особое внимание уделяется роли NEK2 в формировании лекарственной устойчивости: подавление ее активности повышает чувствительность опухолевых клеток к различным цитостатикам. Этот эффект, в частности, связывают со снижением экспрессии ABC-транспортеров и, как следствие, ослаблением эффлюкса химиотерапевтических препаратов из клетки. В совокупности данные механизмы подчеркивают статус NEK2 как ключевого регуляторного узла, одновременно контролирующего клеточное деление, фенотипическую пластичность и реакцию опухоли на терапию [19].
При интерпретации результатов важно учитывать различия между клеточными моделями. В проведенном авторами исследовании первичная культура имела более высокий исходный уровень экспрессии NEK2 и более выраженный ответ на ее нокдаун, что соответствует концепции «онкогенной зависимости», при которой опухолевые клетки особенно чувствительны к подавлению гиперэкспрессированных генов-драйверов [7]. Эта культура была получена от пациента с низкодифференцированной ацинарной аденокарциномой предстательной железы (Глисон 7: 4+3), что указывает на более агрессивное течение заболевания и может отражать обратную связь между уровнем NEK2 и степенью дифференцировки опухоли [5].
Различия эффектов также связаны с происхождением моделей: PC-3 имеет черты мелкоклеточной нейроэндокринной карциномы, DU145 получена из метастаза в ЦНС, тогда как первичная культура лучше сохраняет характеристики локальной опухоли и ее микроокружения. Эти особенности, вероятно, определяют как уровень базальной экспрессии NEK2, так и выраженность фенотипических изменений при ее ингибировании [20, 21].
Хотя РНК-интерференция с синтетическими миРНК является стандартным методом, полностью исключить off-target-эффекты невозможно, поэтому часть наблюдаемых эффектов может быть опосредована влиянием на родственные гены и сигнальные пути. Для более точного разграничения on-target и off-target-эффектов необходимы транскриптомные исследования и подтверждение результатов альтернативными подходами, такими как CRISPR/Cas9 или фармакологические ингибиторы. Отсутствие анализа клеточного цикла, апоптоза и экспрессии про- и антиапоптотических генов ограничивает глубину механистических выводов [10].
Работа выполнена в условиях in vitro, что не учитывает трехмерную организацию опухоли, влияние стромы, гипоксии и внутритканевой гетерогенности. Кроме того, использование культуры от одного пациента затрудняет экстраполяцию данных на широкую популяцию, поэтому для надежных выводов о роли NEK2 в низкодифференцированном РПЖ требуются исследования на большем числе образцов.
Для трансляции полученных результатов в клиническую практику необходимы доклинические эксперименты in vivo, разработка специфических низкомолекулярных ингибиторов NEK2 и определение подгрупп пациентов, которые могут получить наибольшую пользу от такой терапии, прежде всего с высокой экспрессией этой киназы. Перспективным является изучение комбинированных схем лечения с гормональной терапией и химиопрепаратами для оценки синергии и преодоления лекарственной резистентности.
Дальнейшие исследования должны включать расширенный набор первичных культур и органоидных моделей, а также анализ влияния NEK2 на андроген-зависимую сигнализацию, учитывая роль альтернативных путей активации андрогенового рецептора в прогрессии кастрационно-резистентного РПЖ [20–22]. Параллельно важно развивать направление малых молекул-ингибиторов NEK2 как более практичную альтернативу миРНК-подходам и проводить крупные клинические исследования, связывающие уровень экспрессии NEK2 с прогнозом и ответом на терапию [7].
Таким образом, стратификация пациентов по уровню экспрессии NEK2 может иметь существенное клиническое значение, однако ее внедрение возможно только после подтверждения в масштабных проспективных исследованиях с анализом клинических исходов.
Заключение
Данное исследование демонстрирует, что серин-треониновая киназа NEK2 является критической для жизнеспособности и миграционной активности клеток РПЖ. Функциональное подавление NEK2 с помощью РНК-интерференции приводит к значительному снижению жизнеспособности клеток (в 2–2,46 раза) и ингибированию миграции во всех трех изученных моделях, включая первичную культуру. Эти результаты расширяют понимание роли NEK2 в онкобиологии РПЖ и подтверждают ее потенциал как терапевтической мишени. Однако трансляция этих находок в клиническую практику требует дополнительных доклинических и клинических исследований, включая валидацию in vivo, разработку специфических ингибиторов и определение клинических предикторов ответа на воздействие на NEK2. Учитывая быстрый прогресс в области ингибирования киназ и растущее понимание механизмов лекарственной устойчивости при РПЖ, NEK2 представляет собой многообещающую мишень для будущих комбинированных терапевтических стратегий.
Conflict of interest
Financing
Библиографическая ссылка
Казанчян Р.О., Головинов И.В., Каплиева И.В., Галина А.В., Ходакова Д.В., Шульга А.А., Гурова С.В., Шевченко А.Н., Комарова Е.Ф., Димитриади С.Н., Андрейко Е.А. ОЦЕНКА ВЛИЯНИЯ НОКДАУНА ГЕНА NEK2 НА ЖИЗНЕСПОСОБНОСТЬ И МИГРАЦИЮ КЛЕТОК РАКА ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ // Современные проблемы науки и образования. 2026. № 2. ;URL: https://science-education.ru/en/article/view?id=34467 (дата обращения: 07.07.2026).
DOI: https://doi.org/10.17513/spno.34467