


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
DEVELOPMENTAL SPEECH DELAY AND AUTISM IN AN INFANT WITH CHROMOSOME 18P DELETION: PRIORITIZING CANDIDATE PROCESSES

Делеции короткого плеча хромосомы 18, или моносомия 18p – это хромосомный синдром, связанный с полной или частичной потерей короткого плеча хромосомы 18. Впервые данную делецию описала группа ученых во главе с Жаном де Груши в 1963 г. [1]. Распространенность синдрома оценивается как 1:50 000 новорожденных детей, соотношение полов составляет 2:3 (М:Ж) [2, 3]. Большинство случаев (более 75%) являются результатом делеции de novo. В остальных случаях изменения генома наследуются от родителей – носителей сбалансированных структурных хромосомных перестроек с участием хромосомы 18 [3]. Изначально при данной хромосомной патологии у детей отмечали небольшой рост, лицевые микроаномалии (круглое лицо, птоз, диспластичные ушные раковины, тонкие губы, опущенные уголки рта, длинный фильтрум) и нарушения психики. С увеличением числа описанных случаев делеции 18p был расширен перечень фенотипических проявлений при этом синдроме, который на данный момент включает микроцефалию, голопрозэнцефалию, клинодактилию, дистонию, короткую шею, аномалии зубов и пороки сердечно-сосудистой системы [4]. Данная делеция является синдромной и индексированной в базе данных Online Mendelian Inheritance in Man [OMIM:146390]. В литературе опубликовано несколько сотен клинических случаев подобных делеций. Выделяют два основных клинических варианта данного синдрома: с грубыми пороками «аринэнцефалической системы» (от циклопии до аринэнцефалии, этот вариант довольно редок) и с отсутствием пороков мозга. Клинические проявления обычно связаны с размером потерянного участка хромосомы 18, а именно с генами, локализованными в области делеции. Необходимо также учитывать сочетанные геномные аномалии, которые могут вносить свой вклад в развитие фенотипа [5, 6, 7]. Одной из ключевых задач при исследовании синдрома делеции 18p является определение генов, потеря которых влечет за собой развитие клинических проявлений. В различных работах был определен широкий спектр генов-кандидатов: CETN1, TGIF1, LAMA1, GNAL, AFG3L2, PTPN2, SMCHD1, TWSG1, DLGAP1, LCCR30, ANKRD12, IMPA2. Изменения в этих генах связывают с психическими и неврологическими нарушениями, заболеваниями желудочно-кишечного тракта, почек и бесплодием [8]. Несмотря на то что данный синдром достаточно подробно исследован, работ, посвященных изучению механизмов развития психических нарушений при делеции 18p, практически нет. В связи с этим определение молекулярных механизмов развития заболевания как при данной делеции, так и при хромосомных аномалиях в целом является актуальной задачей современной медицинской генетики [9, 10, 11]. Понимание механизмов развития патологии при делеции 18p может внести значимый вклад в разработку персонализированного лечения для данной группы пациентов.
В настоящей работе мы представляем результаты клинического, цитогенетического, молекулярно-цитогенетического и биоинформатического исследований делеции короткого плеча хромосомы 18 (18pter-p11.1) у ребенка 10 лет.
Целью настоящей работы являлось формирование геномной сети психических нарушений при синдромальной делеции 18p на основании молекулярно-цитогенетического и биоинформатического исследований хромосомной патологии у ребенка с нарушением психики и микроаномалиями развития.
Материалы и методы исследования
В настоящей работе описывается мальчик 10 лет с задержкой психического развития, расстройством аутистического спектра, задержкой роста и микроаномалиями развития.
Цитогенетическое исследование. Препараты метафазных хромосом получали путем фиксации лимфоцитов периферической крови после культивирования in vitro в соответствии со стандартной методикой [12]. Цитогенетический анализ проводился при помощи дифференциального окрашивания хромосом по длине (GTG-и CBG-окрашивание) по общепринятым протоколам под световым микроскопом при увеличении х1125.
Молекулярно-цитогенетическое исследование. Молекулярно-цитогенетическое исследование проводилось с использованием SNP/олигонуклеотидной микроматрицы Affymetrix Cytoscan HD, содержащей 2 696 550 проб. Исследование методом молекулярного кариотипирования проводили согласно ранее описанному протоколу [13].
Биоинформатический анализ проводился с использованием оригинальной биоинформатической технологии, включающей в себя изучение транскриптомных, метаболомных и интерактомных данных [14], с целью приоритезации генов и процессов-кандидатов, а также определения корреляций и генотип-фенотип.
Исследование было одобрено этическим комитетом ОСП «Научно-исследовательский клинический институт педиатрии и детской хирургии им. академика Ю.Е. Вельтищева» ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России.
Результаты исследования и их обсуждение
При исследовании ребенка были выявлены задержка психического развития, РАС, агрессивное поведение, задержка роста и микроаномалии развития (гипертелоризм глаз и сосков, деформированные ушные раковины, опущенные углы рта, тонкие губы, короткая шея, неполная кожная синдактилия I–II пальцев кистей).
В результате проведенного цитогенетического исследования у ребенка была обнаружена делеция короткого плеча хромосомы 18 (рис. 1). Кариотип ребенка после проведения стандартного цитогенетического исследования: 46,XY,del(18)(p11.1).
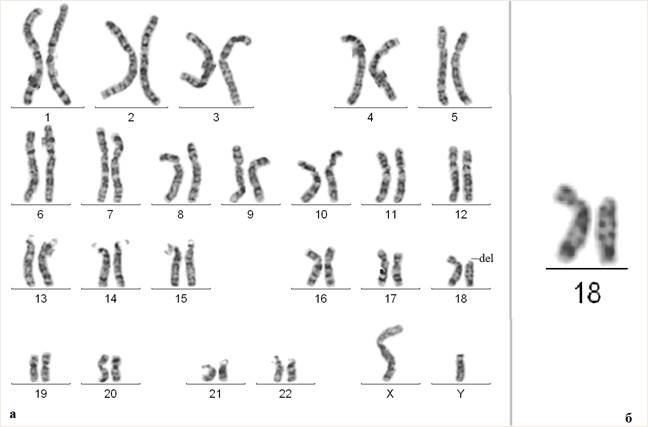
Рис. 1. Результаты цитогенетического исследования ребенка с делецией хромосомы 18, затронувшей участок 18pter-p11.1. На рисунке (а) представлен кариотип пациента, на рисунке (б) – отдельно хромосомы 18 (слева хромосома без изменений, справа –хромосома 18 с делецией короткого плеча)
Для уточнения точек разрыва и размера потерянного участка хромосомы 18, а также поиска других возможных структурных вариаций генома ребенку было проведено молекулярно-цитогенетическое исследование методом молекулярного кариотипирования (SNP array) с последующим биоинформатическим анализом. Исследование подтвердило наличие делеции, затрагивающей участок 18p11.32p11.21, размер которой составил примерно 15 млн пн (рис. 2). Результаты молекулярного кариотипирования согласно ISCN 2016: arr[CRCh37] 18p11.32p11.21(136226_15154053)×1.
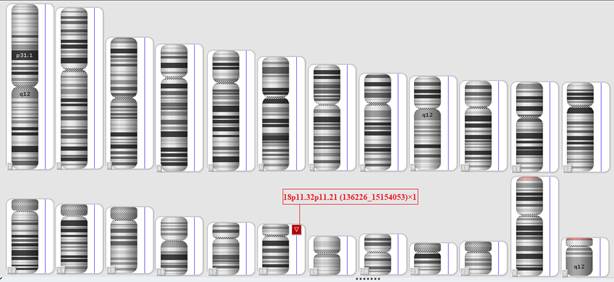
Рис. 2. Результаты молекулярного кариотипирования: делеция 18p11.32p11.21 (отмечена красным цветом)
В дальнейшем был проведен биоинформатический анализ для приоритизации генов и процессов-кандидатов [15, 16, 17]. При этом использовались базы данных и ресурсы OMIM, NCBI Gene, UCSC Genome Browser, String, BioGPS, Gene Ontology, KEGG. В области делеции хромосомы 18 локализовано 115 генов, 57 из которых индексированы в базе данных OMIM, согласно программе анализа данных Chromosome Analysis Suite (ChAS), версия сборки генома (GRCh37/hg19). На первом этапе оценки вклада роли отдельных генов (локализованных в области делеции) в формирование фенотипических проявлений в описываемом случае был проведен анализ литературы и ассоциаций изменений в генах с клиническими проявлениями. Из 57 генов, индексированных в OMIM и расположенных в делетированном участке 18p11.32p11.21, 11 генов ассоциированы с описанными ранее заболеваниями (табл. 1): SMCHD1 [OMIM:614982], LPIN2 [OMIM:605519], TGIF1 [OMIM:602630], LAMA1 [OMIM:150320], NDUFV2 [OMIM:600532], APCDD1 [OMIM:607479], PIEZO2 [OMIM:613629], GNAL [OMIM:139312], TUBB6 [OMIM:615103], AFG3L2 [OMIM:604581], MC2R [OMIM:607397].
Таблица 1
Гены, расположенные в делетированном участке 18p11.32p11.21, индексированные в OMIM и ассоциированные с известными клиническими проявлениями (данные извлечены из базы данных OMIM за март 2022 г).
|
Хромосомная локализация |
Ген |
Заболевания |
Тип наследования |
|
SMCHD1 |
Синдром BAMS (Босма-архинии микрофтальмии) |
Аутосомно-доминантный |
|
|
Плече-лопаточно-лицевая дистрофия |
Дигенно-доминантный |
||
|
18p11.31 |
LPIN2 |
Синдром Маджида |
Неизвестно |
|
18p11.31 |
Голопрозэнцефалия |
Аутосомно-доминантный |
|
|
18p11.31 |
LAMA1 |
Синдром Поретти–Болтсхаузер |
Аутосомно-рецессивный |
|
18p11.22 |
NDUFV2 |
Недостаточность митохондриального комплекса I, тип 7 |
Аутосомно-рецессивный |
|
18p11.22 |
APCDD1 |
Гипотрихоз |
Аутосомно-доминантный |
|
18p11.22-p11.21 |
PIEZO2 |
Синдром Мардена–Уокера |
Аутосомно-доминантный |
|
Дистальный артрогрипоз, тип 3 |
Аутосомно-доминантный |
||
|
Дистальный артрогрипоз, тип 5 |
Аутосомно-доминантный |
||
|
Дистальный артрогрипоз с нарушением проприоцепции и осязания |
Аутосомно-рецессивный |
||
|
18p11.21 |
GNAL |
Дистония-25 |
Аутосомно-доминантный |
|
18p11.21 |
TUBB6 |
Паралич лицевого нерва, конгенитальный, с птозом и велофарингеальной дисфункцией |
Аутосомно-доминантный |
|
18p11.21 |
AFG3L2 |
Оптическая нейропатия-12 |
Аутосомно-доминантный |
|
Спастическая атаксия-5, аутосомно-рецессивная |
Аутосомно-рецессивный |
||
|
Спиноцеребеллярная атаксия 28 |
Аутосомно-доминантный |
||
|
18p11.21 |
MC2R |
Глюкокортикоидная недостаточность вследствие невосприимчивости к АКТГ (семейная глюкокортикоидная недостаточность) |
Аутосомно-рецессивный |
По данным литературы, все представленные в таблице 1 заболевания или их признаки встречались у пациентов с делецией 18p [6, 7]. У исследуемого пациента ни один из моногенных синдромом, обозначенных выше, не был диагностирован. Однако у ребенка, так же как и при некоторых из данных состояний, присутствовали когнитивные нарушения. В данном случае особый интерес при корреляции генотип/фенотип для описываемого ребенка представляли гены GNAL и AFG3L2, аномалии в которых могут приводить к развитию двигательных нарушений. Изменения в генах NDUFV2 и LAMA1 были ассоциированы с развитием психических нарушений и гипотонии мышц, а в гене PIEZO2 – с неврологическими нарушениями, гипотонией, микроаномалиями развития [18, 19]. Следующим этапом биоинформатического анализа при приоритизации генов-кандидатов являлся анализ экспрессии генов [14, 20]. Фильтрация генов производилась по уровню их экспрессии в клетках центральной нервной системы. В ходе оценки данных экспрессии 57 генов, локализованных в делетированном участке (согласно данным BioGPS (GeneAtlas U133A) [21]), было установлено, что повышенную экспрессию в различных областях головного мозга имеют 9 генов: EPB41L3 [OMIM:605331], PTPRM [OMIM:176888], MTCL1 [OMIM:615766], RAB31 [OMIM:605694], GNAL [OMIM:139312], SEH1L [OMIM:609263], LDLRAD4 [OMIM:606571], RNMT [OMIM:603514], ANKRD12 [OMIM:610616]. На рисунке 3 представлены локализация генов в коротком плече хромосомы 18, а также участки мозга, в которых они имеют повышенную экспрессию относительно других тканей.
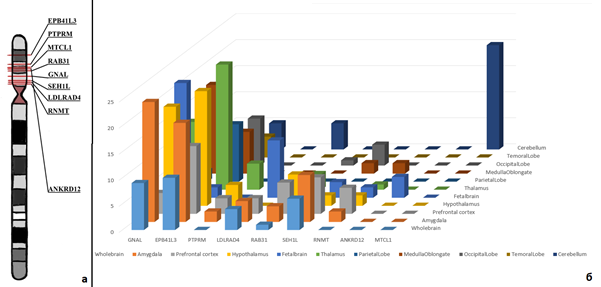
Рис. 3. (а) Локализация генов, расположенных в коротком плече хромосомы 18 и имеющих повышенную экспрессию в головном мозге; (б) профили экспрессии делетированных генов в различных областях головного мозга, полученные с помощью ресурса BioGPS [21]
Дополнительным параметром при приоритизации генов-кандидатов являлись их функциональная значимость и участие в значимых биологических процессах. В настоящем исследовании, в первую очередь, учитывались процессы, которые, согласно данным Киотской энциклопедии генов и геномов KEGG [22], являются ответственными за развитие и функционирование головного мозга. В этом контексте среди делетированных генов особый интерес представляют гены NDUFV2, DLGAP1, GNAL, TUBB6, участвующие в таких процессах, как аксональное наведение, функционирование глутаматергических и допаминергических синапсов, а также в процессах, приводящих к развитию нейродегенеративных заболеваний [23, 24]. Таким образом, на основании анализа литературных данных и ассоциаций с заболеваниями с использованием OMIM было приоритезировано 5 генов, на основании данных об экспрессии с использованием BioGPS –9 генов, и еще 4 гена были приоритезированы на основании данных об их функциональной значимости с использованием KEGG. При этом некоторые гены были ассоциированы с психическими заболеваниями на основании сразу нескольких показателей. Например, ген GNAL имеет повышенную экспрессию в клетках головного мозга, участвует в функционировании дофаминергического синапса. По данным литературы, аномалии гена GNAL связывают с развитием психических нарушений. Ген NDUFV2 задействован в процессе нейродегенерации и, по данным литературы, аномалии данного гена связывают с развитием психических нарушений и гипотонией мышц. В результате трех основных этапов биоинформатического исследования всего было определено 15 генов-кандидатов (NDUFV2, DLGAP1, TUBB6, EPB41L3, PTPRM, MTCL1, RAB31, SEH1L, LDLRAD4, RNMT, ANKRD12, GNAL, AFG3L2, LAMA1, PIEZO2), изменения в которых могут способствовать возникновению нарушений функционирования и развития головного мозга и, как следствие, развитию психических нарушений у исследуемого пациента [25]. На этапе интерактомного анализа все гены-кандидаты были объединены в геномную сеть заболевания и представлены на рисунке 4.
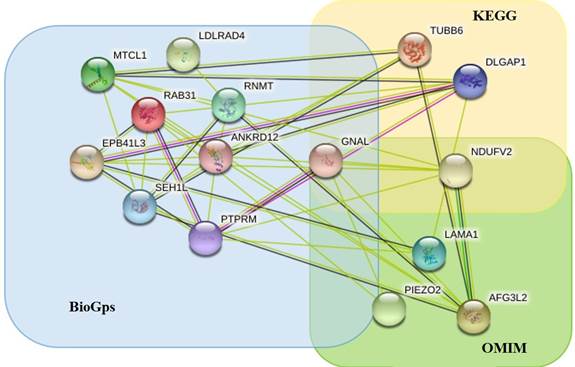
Рис. 4. На рисунке представлен интерактом генов-кандидатов, созданный с использованием STRING (подробнее см. https://stringdb.org [26]). Гены, имеющие повышенную экспрессию в мозге, находятся в зоне голубого квадрата (BioGps); гены, участвующие в процессах, связанных с развитием и функционированием мозга, – в желтом квадрате (KEGG); гены, ассоциированные с психическими заболеваниями, – в зоне зеленого квадрата (OMIM). Нахождение в той или иной зоне говорит о причине приоритизации гена
Суммируя полученные данные, можно сделать выводы о том, что вариации числа копий последовательностей ДНК вышеописанных генов могут негативно сказываться на развитии и функционировании центральной нервной системы, приводя к развитию фенотипических проявлений у описываемого индивидуума. Использование оригинального алгоритма для приоритизации генов-кандидатов позволило полноценно изучить механизмы развития нервно-психических нарушений у пациента. Полученные результаты могут быть фундаментом для формирования персонализированных рекомендаций по ведению пациентов с хромосомными аномалиями и повышения уровня медико-генетического консультирования подобных пациентов.
Заключение
В данной работе представлен случай синдромальной делеции короткого плеча хромосомы 18 у ребенка с расстройством аутистического спектра, психомоторными нарушениями и микроаномалиями развития. Были проведены цитогенетическое и молекулярно-цитогенетические исследования для точного определения объема потерянного генетического материала. С помощью оригинальной биоинформатической технологии были определены гены в участке делеции, оказывающие наиболее значимое влияние на развитие психических нарушений у ребенка. Данные гены были объединены в персонифицированную геномную сеть психических нарушений при делеции 18p11.32p11.21. Геномная сеть состояла из 15 генов: NDUFV2, DLGAP1, TUBB6, EPB41L3, PTPRM, MTCL1, RAB31, SEH1L, LDLRAD4, RNMT, ANKRD12, GNAL, AFG3L2, LAMA1, PIEZO2. Полученные данные о механизмах патогенеза данной хромосомной патологии могут быть использованы для разработки тактики ведения пациента и персонифицированной терапии. Описываемый случай хромосомной аномалии демонстрирует необходимость применения молекулярно-цитогенетических методов исследования с дальнейшим биоинформатическим анализом с целью определения механизмов возникновения геномных перестроек [27] и формирования индивидуальной геномной сети клинических проявлений и/или механизмов заболевания [10, 14].
Библиографическая ссылка
Васин К.С., Ворсанова С.Г., Колотий А.Д., Куринная О.С., Зеленова М.А., Кравец В.С., Смирнова О.А., Курамагомедова Р.Г., Юров И.Ю. ЗАДЕРЖКА ПСИХОРЕЧЕВОГО РАЗВИТИЯ И РАССТРОЙСТВО АУТИСТИЧЕСКОГО СПЕКТРА У РЕБЁНКА С ДЕЛЕЦИЕЙ КОРОТКОГО ПЛЕЧА ХРОМОСОМЫ 18: ПРИОРИТИЗАЦИЯ ПРОЦЕССОВ-КАНДИДАТОВ // Современные проблемы науки и образования. 2022. № 5. ;URL: https://science-education.ru/en/article/view?id=32135 (дата обращения: 02.08.2026).
DOI: https://doi.org/10.17513/spno.32135