


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
STUDY OF 4-OXOPIRIMIDINE DERIVATIVES INTERACTION WITH THE ACTIVE SITE OF CYCLOOXYGENASE-2 USING MOLECULAR DYNAMICS

Цель исследования
Определить возможность связыванияпрогнозируемых производных 4-оксопиримидина с аминокислотами активного центра циклооксигеназы-2 (ЦОГ-2) и выявить из них наиболее перспективные ингибиторы.
Методы исследования
Основной фармакологический эффект НПВС заключается в ингибировании циклооксигеназы (ЦОГ) и липооксигеназы (ЛОГ) – ключевых ферментов метаболизма арахидоновой кислоты. Циклооксигеназа-1 (ЦОГ-1) продуцируется в обычных условиях и регулирует образование в организме простаноидов. Продукция циклооксигеназы-2 в значительной степени индуцируется процессом воспаления. Исходя из этого, наибольший интерес вызывает поиск избирательных ингибиторов ЦОГ-2. Селективное ингибирование ЦОГ-2 обеспечивает противовоспалительный эффект, снижает вероятность развития многих побочных проявлений, связанных с ингибированием физиологического (не связанного с воспалением) биосинтеза простаноидов[5].
ЦОГ-2 является гомодимером, каждая из субъединиц одновременно активна и содержит циклооксигеназный активный центр (рис.1) [6,7]. Каталитический домен
является основной частью мономера ЦОГ-2 и центром связывания лигандас ферментом.
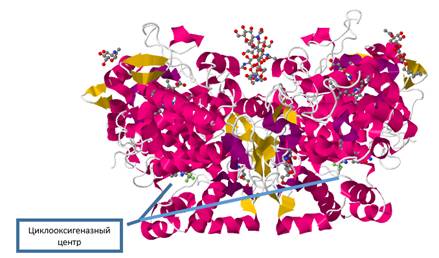
Рис. 1. Строение ЦОГ-2(PDBID: 3NTG) [9]
В качестве инструмента для поиска структур биологически активных веществ был использован метод молекулярной динамики, позволяющий достичь высокой достоверности результатов прогнозирования биологической активности, благодаря детальному описанию молекулярной системы. Объектом моделирования является система «лиганд-фермент». При этом, в процессе вычислительного эксперимента записываются различные параметры образующегося комплекса, такие как энергия взаимодействия между лигандом и ферментом, дистанции между отдельными группами атомов. Моделирование молекулярной динамики проводилось с помощью программы БИОЭВРИКА. В качестве биологической мишени использована молекула фермента ЦОГ-2, трехмерная структура которой была получена методом рентгеноструктурного анализа(PDBID: 4M11) [8]. К молекуле фермента в соответствии с валентностями были добавлены атомы водорода. Макромолекула была помещена в водное окружение. Далее проводилось оптимизация геометрии системы.Термостатированиеосуществлялось с использованием термостата Берендсена при температуре 300К, длительность моделирования составила 512 нс (Рис.2).

Рис.2. Молекулярная система «ЦОГ-2 – лиганд – вода»
При моделировании лиганд помещался в растворителе у входа в канал активного центра. Такое начальное расположение лиганда позволяет определить возможность его проникновения в каталитический центр фермента. В процессе моделирования оценивалась энергия ван-дер-ваальсова взаимодействия как показатель связывания лиганда с аминокислотами активного центра. На основе этих данных оценивалась длительность связывания лиганда с ферментом.
Результаты исследования и их обсуждение
В качестве исследуемых веществ были выбраны три производных 4-оксопиримидина, химические структуры которых приведены в таблице 1.
Таблица 1
Химические структуры и лабораторные шифры исследуемых веществ
|
Шифр исследуемых соединений |
Структура |
|
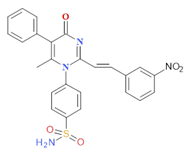
PMSmNO2 |
|
|
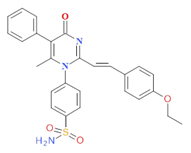
PMSpEtO |
|
|
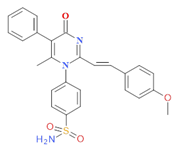
PMSpMeOB |
|
По результатам моделирования была проведена оценка длительности связывания исследуемых веществ с активным центром фермента ЦОГ-2 (таблица 2).
Таблица 2
Оценка длительности связывания исследуемых веществ с активным центром ЦОГ-2
|
Исследуемые вещества |
Длительность связывания, нс |
|
PMSmNO2 |
84,21 |
|
PMSpEtO |
56,89 |
|
PMSpMeOB |
46,84 |
Моделирование молекулярной динамики показывает, что время связывания каждого из исследуемых веществ составляет не менее 40 нс, что доказывает возможность их связывания с аминокислотами катилитического центра ЦОГ-2.
Выводы
Результаты вычисления показывают, что исследуемые вещества способны проникать в активный центр ЦОГ-2 из водного окружения. Наибольшей активностью, предположительно, обладает вещество PMSmNO2, что возможно связано с наличием нитро-группы в фенильном радикале. Полученные результаты представляют интерес для дальнейшего контсруирования новых НПВС в ряду производных 4-оксопиримидина и позволяют предположить наличие у исследуемых веществ противовоспалительной активности.
Рецензенты:Компанцев В.А.,д.фарм.н.,профессор кафедры неорганической, физической и коллоидной химии Пятигорского медико-фармацевтического института — филиала ГБОУ ВПО ВолгГМУ Минздрава России, г.Пятигорск;
Лазарян Д.С., д.фарм.н.,заведующий кафедрой фармацевтической и токсикологической химии, профессорПятигорского медико-фармацевтического института — филиала ГБОУ ВПО ВолгГМУ Минздрава России, г.Пятигорск.
Библиографическая ссылка
Гендугов Т.А., Щербакова Л.И., Глушко А.А., Кодониди И.П., Сочнев В.С. ИЗУЧЕНИЕ ВЗАИМОДЕЙСТВИЯ ПРОИЗВОДНЫХ 4-ОКСОПИРИМИДИНА С АКТИВНЫМ ЦЕНТРОМ ЦИКЛООКСИГЕНАЗЫ-2 МЕТОДОМ МОЛЕКУЛЯРНОЙ ДИНАМИКИ // Современные проблемы науки и образования. 2015. № 2-2. ;URL: https://science-education.ru/en/article/view?id=22796 (дата обращения: 01.07.2026).