



Одним из самых распространенных заболеваний в наши дни является язвенная болезнь желудка и двенадцатиперстной кишки. Независимо от этиологии основным механизмом развития данного заболевания является увеличение продукции соляной кислоты и ее патологическое влияние на слизистую ЖКТ [4; 9].
С целью расширения ассортимента препаратов для лечения язвы желудка нами был разработан новый комбинированный лекарственный препарат, сочетающий в себе противоязвенное и ранозаживляющее действия [5; 6] и обладающий улучшенными биофармацевтическими свойствами [1] – липосомальный гель с фамотидином и маслом кедровым.
Целью настоящей работы является разработка методик анализа действующих компонентов липосомального геля.
Оценку качества разработанного геля проводили в соответствии с требованиями ГФ XII по таким показателям, как органолептические свойства, степень однородности, рН, подлинность и количественное содержание фамотидина.
Для проведения испытаний на подлинность фармакологически активных компонентов геля готовили три модельные смеси, содержащие:
1) фамотидин и кедровое масло;
2) только фамотидин;
3) только кедровое масло.
В кедровом масле основными БАВ являются токоферолы. Для их идентификации использовали химические и физико-химические методы определения.
В качестве физико-химического метода идентификации токоферолов использовали метод ТСХ на пластинках «Сорбфил». Токоферолы экстрагировали спиртом этиловым. На линию старта наносили спиртовые извлечения исследуемого геля, масла кедрового и спиртовой раствор фамотидина. Пластинку после подсушивания пятен помещали в хроматографическую камеру с системой растворителей н-гексан – бензол – хлороформ (70:15:15). После опрыскивания пластинки концентрированной азотной кислотой наблюдали появление пятен красно-оранжевого цвета на одном уровне только в модельных смесях 1 и 3.
Кроме метода ТСХ, присутствие токоферолов в липосомальном геле подтверждали химическими реакциями. При нагревании водного раствора геля с концентрированной азотной кислотой наблюдали образование токоферилхинонов, окрашенных в красно-оранжевый цвет. При взаимодействии с феррицианидом (III) калия в щелочной среде наблюдается образование продуктов, окрашенных в ярко-желтый цвет [3].
Специфичность реакций подтверждали также на трех модельных смесях, положительные реакции наблюдали в смесях 1 и 3.
Для идентификации фамотидина в геле были разработаны методики его определения химическими реакциями и спектрофотометрическим методом.
Изучение УФ-спектров поглощения фамотидина и кедрового масла показало, что наличие БАВ в масле оказывает влияние на оптическую плотность фамотидина. Поэтому подтверждение подлинности и количественное определение фамотидина в геле проводили после предварительного разделения компонентов. Около 2,5 г геля (точная навеска) растворяли в воде, смешивали с 10 мл хлороформа для экстракции кедрового масла из ЛП. Раствор помещали в делительную воронку, хлороформное извлечение сливали. Экстракцию проводили еще дважды 10 мл и 5 мл хлороформа до отсутствия положительной реакции на токоферолы. Водную фазу фильтровали в мерную колбу вместимостью 50 мл и доводили объем водой до метки. 3 мл полученного раствора смешивали с 10 мл ацетатного буфера с pH 4,5 в мерной колбе вместимостью 50 мл, объем полученной смеси доводили до метки спиртом этиловым 96%. Оптическую плотность полученного раствора измеряли на спектрофотометре в области 220-320 нм относительно раствора сравнения, приготовленного по следующей методике: 13 мл ацетатного буфера с pH 4,5 помещали в мерную колбу вместимостью 50 мл, доводили объем до метки спиртом этиловым 96%. Спектр поглощения раствора испытуемого образца в смеси этанол – ацетатный буфер характеризуется наличием одного максимума поглощения при длине волны 270 нм, что подтверждается данными литературных источников [7; 8].
Для идентификации фамотидина были также разработаны методики качественного анализа с использованием химических реакций. Предложена реакция разложения фамотидина при нагревании с раствором натрия гидроксида. Выделившийся аммиак обнаруживали по посинению влажной красной лакмусовой бумаги. Так как фамотидин является органическим основанием, использовали реакцию образования ионного ассоциата с эозином. После экстракции хлороформом наблюдали красно-оранжевое окрашивание хлороформного слоя. Для определения специфичности предложенных реакций использовали три вышеуказанные модельные смеси. Положительные реакции наблюдались в смесях 1 и 2. Это свидетельствует об отсутствии взаимодействия реактивов с компонентами кедрового масла. Полученные данные подтверждают специфичность методик качественного определения компонентов в геле.
Метод спектрофотометрии использовался нами и для количественного определения фамотидина в полученном геле. Испытуемый образец и раствор сравнения готовили по методике, описанной выше. Оптическую плотность испытуемого раствора измеряли на спектрофотометре при длине волны 270 нм относительно раствора сравнения. Параллельно измеряли оптическую плотность раствора стандартного образца. Рабочий стандартный образец (РСО) готовили следующим образом. 0,02 г фамотидина СО помещали в мерную колбу вместимостью 100 мл, растворяли и доводили до метки смесью спирт этиловый 96% – ацетатный буфер в соотношении 4:1. Затем 5 мл полученного раствора переносили в мерную колбу вместимостью 100 мл, объем доводили до метки вышеуказанной смесью. Спектры поглощения стандартного и испытуемого образцов в смеси этанол – ацетатный буфер характеризуются наличием одного максимума при длине волны 270 нм (рис. 1).
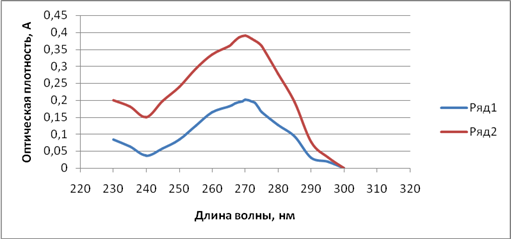
Рис. 1. Спектры поглощения испытуемого и стандартного образцов
Ряд 1 –УФ-спектр стандартного образца фамотидина
Ряд 2 – УФ-спектр испытуемого геля
В связи с современными требованиями разработанные методики анализа должны подвергаться валидации по показателям: специфичность, линейность, прецизионность, правильность, предел обнаружения [2].
Для определения линейности готовили серию растворов СО фамотидина с различной концентрацией и измеряли оптическую плотность при длине волны 270 нм. По полученным данным (табл. 1) строили градуировочный график (рис. 2), рассчитывали уравнение графика и коэффициент корреляции.
Таблица 1 – Результаты определения линейности методики спектрофотометрического определения фамотидина в геле
|
Объем стандартного раствора, мл |
С, мг/100 мл (xi) ·10-4 |
Оптическая плотность, А (yi) |
xi*yi ·10-4 |
xi2 ·10-8 |
yi2 |
|
1 |
0,0400 |
0,188 |
0,0075 |
0,0016 |
0,0353 |
|
2 |
0,0800 |
0,344 |
0,0275 |
0,0064 |
0,1183 |
|
3 |
0,1200 |
0,505 |
0,0606 |
0,0144 |
0,2550 |
|
4 |
0,1600 |
0,661 |
0,1058 |
0,0256 |
0,4363 |
|
5 |
0,2000 |
0,818 |
0,1636 |
0,0400 |
0,6691 |
|
6 |
0,2400 |
0,978 |
0,2347 |
0,0576 |
0,9565 |
|
å |
0,8400 |
3,494 |
0,5204 |
0,1456 |
2,4706 |
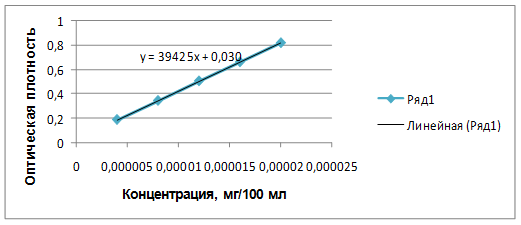
Рис. 2. Градуировочный график зависимости оптической плотности раствора фамотидина от концентрации.
В результате эксперимента было установлено, что в данной области концентраций график (рис. 2) имеет линейный характер и описывается уравнением у=39425х+0,0301. Коэффициент корреляции равен 0,9999, и пересечение с осью Y не более 2% от величины коэффициента а, что позволяет использовать данную методику для количественного определения фамотидина в геле.
Для определения прецизионности использовали приготовленный нами гель с точным содержанием компонентов. Для этого проводили 9 параллельных определений на трех уровнях концентраций.
Путем статистической обработки полученных результатов рассчитывали стандартное отклонение (SD) и относительное стандартное отклонение (RSD) (табл. 2).
Таблица 2 – Результаты определения прецизионности количественного определения фамотидина спектрофотометрическим методом
|
№ п/п |
Х, % |
Хi-Хср |
(Хi-Хср)2 |
SD=0,821 RSD=0,822% |
|
1 |
101,12 |
1,2322 |
1,5184 |
|
|
2 |
101,25 |
1,3622 |
1,8556 |
|
|
3 |
100,21 |
0,3222 |
0,1038 |
|
|
4 |
99,9 |
0,0122 |
0,0002 |
|
|
5 |
99,67 |
-0,2178 |
0,0474 |
|
|
6 |
99,34 |
-0,5478 |
0,3001 |
|
|
7 |
99,12 |
-0,7678 |
0,5895 |
|
|
8 |
99,21 |
-0,6778 |
0,4594 |
|
|
9 |
99,17 |
-0,7178 |
0,5152 |
|
|
Хср |
99,89 |
Σ(Хi-Хср)2 |
5,3896 |
Таким образом, в ходе эксперимента было установлено, что в данной методике величина RSD менее 1%, что характеризует надежность анализа в выбранных условиях.
Для тестирования методики на правильность готовили модельные смеси с точным содержанием определяемого вещества. Далее по разработанным методикам анализа было проанализировано 9 образцов на трех уровнях концентраций в пределах аналитической области.
По полученным данным рассчитывали количественное содержание фамотидина в испытуемом геле и значение открываемости (R) (табл. 3). Полученные значения открываемости соответствуют рекомендациям AOAC (среднее значение открываемости должно находиться в пределах 97-103%).
Таблица 3 – Результаты определения правильности методики количественного определения фамотидина спектрофотометрическим методом
|
№ п/п |
R, % |
Ri-Rср |
(Ri-Rср)2 |
SD=0,947 RSD=0,95%
|
|
|
1 |
100,08 |
-0,0378 |
0,0014 |
||
|
2 |
100,1 |
-0,0178 |
0,0003 |
||
|
3 |
100,5 |
0,3822 |
0,1461 |
|
|
|
4 |
101,2 |
1,0822 |
1,1712 |
|
|
|
5 |
99,2 |
-0,9178 |
0,8423 |
|
|
|
6 |
98,12 |
-1,9978 |
3,9911 |
|
|
|
7 |
101,03 |
0,9122 |
0,8321 |
|
|
|
8 |
100,35 |
0,2322 |
0,0539 |
|
|
|
9 |
100,48 |
0,3622 |
0,1312 |
||
|
Rср |
100,12 |
Σ(Ri-Rср)2 |
7,1698 |
По данным, приведенным в таблице 2, определяли предел обнаружения (Cmin= 0,63·10-4г) и предел количественного обнаружения (QL = 2,1·10-5г).
Как показали результаты валидационной оценки, спектрофотометрический метод позволяет с достаточной точностью определить количественное содержание фамотидина в полученном геле.
Выводы
1. Разработаны методики качественного и количественного определения действующих компонентов липосомального геля.
2. Проведена валидация предлагаемых методик анализа по показателям: специфичность, линейность, прецизионность, правильность, предел обнаружения, предел количественного обнаружения.
Рецензенты:
Вергейчик Е.Н., доктор фармацевтических наук, профессор кафедры фармацевтической химии Пятигорского филиала ГБОУ ВПО «Волгоградский государственный медицинский университет» Министерства здравоохранения РФ, г. Пятигорск.
Лазарян Д.С., доктор фармацевтических наук, профессор кафедры токсикологической химии Пятигорского филиала ГБОУ ВПО «Волгоградский государственный медицинский университет» Министерства здравоохранения РФ, г. Пятигорск.