



Введение
Хелатные комплексы редкоземельных элементов (РЗЭ) характеризуются интенсивной люминесценцией, причем у некоторых из них наблюдается триболюминесценция (ТЛ) [1; 2]. Исследования явления ТЛ актуальны как с фундаментальной (преобразование механической энергии в световую), так и с прикладной точки зрения (разработка оптических сенсоров нового поколения для регистрации повреждений в критических объектах).
Одним из перспективных классов триболюминофоров являются комплексные соединения, содержащие лантаниды. Ранее экспериментально были изучены ТЛ-свойства хелатного комплекса европия (III) состава Eu(NO3)3(ГМФА)3 (ГМФА – OP(N(CH3)2)3, гексаметилфосфотриамид) [1].
Целью настоящей работы является теоретическое физико-химическое исследование структуры, электронного строения и спектрального поведения аналога комплекса европия – хелата лантана (III) La(NO3)3(ГМФА)3 (I).
Методы исследования
С помощью программы GAMESS [7] методами функционала плотности DFT и TDDFT с гибридным обменно-корреляционным функционалом PBE0 [6], Штутгартским псевдопотенциалом и базисом ECP-MWB [8] в вакуумном приближении выполнены квантово-химические расчеты строения и свойств кластеров комплекса I в основном и возбужденных состояниях.
Результаты и обсуждение
Известно, что метод DFT лучше классического неэмпирического метода воспроизводит многие физико-химические параметры систем этого класса [3; 4]. Согласно расчетам, модельная геометрическая структура комплекса I согласуется с экспериментальными характеристиками изоструктурного комплекса Eu(NO3)3(ГМФА)3, обладающего ТЛ [1].
Оценка параметров электронного строения кристалла проведена на основе расчетов молекулярных ассоциатов (La(NO3)3ГМФА3)n (n = 1–4) от мономера до тетрамера.
Установлено, что большинство расчетных порядков связей при переходе от экспериментальной к оптимальной геометрии комплекса I изменяются незначительно. При этом порядки связей La-O в экваториально расположенных лигандах NO3 и в полярно расположенных нейтральных лигандах ГМФА заметно уменьшаются, что подтверждается существенным ростом длин этих связей.
Согласно расчетам, данная молекулярная система является сильно полярной, ее дипольный момент равен 9.2 Д. Установлено, что распределение электронной плотности комплекса I при изменении геометрии от оптимальной к экспериментальной изменяется незначительно, но заряд РЗ иона уменьшается 0.22е. При этом дипольный момент молекулы увеличивается на 6.58 Д, что свидетельствует о существенном росте поляризации системы.
Состав граничных молекулярных орбиталей (МО) и энергетические щели между ними позволили определить важные параметры электронного строения молекулярной системы и спрогнозировать вероятные центры возбуждения при переходе комплекса в возбужденное синглетное и мультиплетное состояния. Согласно расчетным данным верхняя занятая МО (ВЗМО) является nN-МО, характеризующей неподеленную электронную пару (НЭП) атома азота лиганда ГМФА (рис. 1), энергия которой изменяется от –6.40 до –5.82 эВ при переходе от мономера к тетрамеру. Нижняя вакантная МО (НВМО) является π*-МО, характеризующей π–систему группы NO3, ее энергия при этом переходе изменяется от –0.82 до –1.06 эВ. Следовательно, энергетическая щель между граничными МО при переходе от мономера к тетрамеру уменьшается от 5.58 до 4.76 эВ.
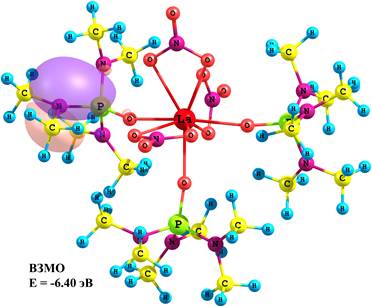
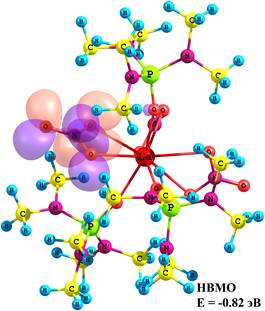
Рис. 1. Энергии и состав граничных МО La(NO3)3ГМФА3
Из состава граничных МО видно, что электронное возбуждение данной системы будет определяться в основном свойствами лигандов, т.е. в электронных спектрах поглощения и люминесценции вероятно появление интенсивных пиков, обусловленных возбуждением электронов лигандов.
Оценка межмолекулярного взаимодействия в кристалле проведена на основе расчетов молекулярных ассоциатов (La(NO3)3ГМФА3)n (n = 2–4) (Рис. 2). Как указано выше, в работе [1] экспериментально изучены ТЛ-свойства комплекса европия (III) Eu(NO3)3(ГМФА)3. Для выявления детального механизма ТЛ в хелатных соединениях состава Ln(NO3)3ГМФА3 (Ln = Eu, Tb) рассмотрен более простой электронный аналог комплекса европия (III) – хелат лантана (III). Выполнено квантово-химическое моделирование влияния смещения слоев молекул относительно друг друга на 0.5, 1.0 и 1.5 ангстрем в кластере (La(NO3)3ГМФА3)4. Определено воздействие дислокации в кристалле на электронное строение кластера. При модельном смещении слоев происходит значительное изменение термодинамических параметров системы, полная энергия тетрамера возрастает на 5.10 эВ. При этом наблюдается значительная поляризация системы, дипольный момент тетрамера возрастает от 0.0 до 7.32 Д и направлен вдоль смещения слоев кластера (рис. 2).
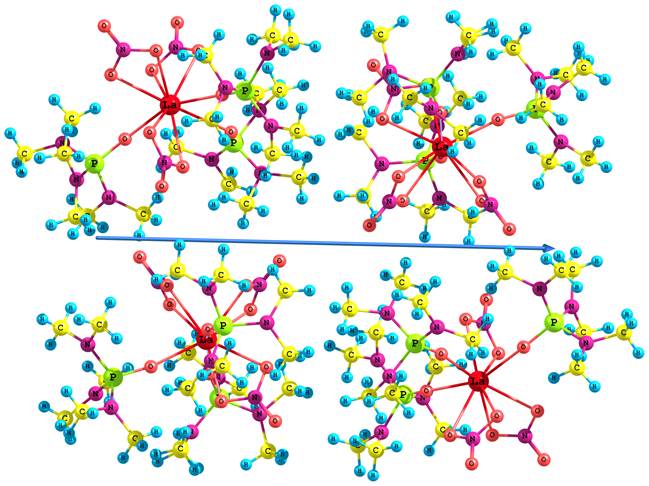
Рис. 2. Модельное смещение слоев молекул в тетрамере (La(NO3)3ГМФА3)4. Стрелкой показано направление дипольного момента.
Таким образом, квантово-химическое моделирование показало, что при механическом воздействии происходит смещение слоев кристалла вдоль плоскостей спайности, приводящее к поляризации системы. Согласно данным расчетов смещение слоев существенно изменяет распределение электронной плотности, состав и энергии граничных МО системы. Это может приводить к возникновению противоположно заряженных плоскостей и появлению ТЛ.
Методом TDDFT/PBE0/ECP-MWB проведен расчет 20 возбужденных синглетных и триплетных состояний комплекса I, смоделирован его УФ-спектр поглощения. Результаты расчетов сопоставлены с экспериментальным спектром поглощения соединения I (рис. 3). Следует учитывать, что экспериментальный спектр получен для раствора хелата, а квантово-химические расчеты проведены для молекул в вакуумном приближении.
Согласно расчетам электронных синглет-синглетных переходов возбуждение молекулярной системы в синглетном состоянии более чем на 90% обусловлено переходами валентных электронов НЭП атомов азота нейтральных лигандов ГМФА на вакантные молекулярные уровни, соответствующие несвязывающей π*-МО π–системы групп NO3. Таким образом, наиболее вероятные центры электронного возбуждения при переходе комплекса I в возбужденные синглетное и мультиплетные состояния локализованы именно на лигандах. На основании этого можно предположить, что широкая полоса с максимальной интенсивностью (λпогл = 300 нм) экспериментального спектра поглощения комплекса I обусловлена внутрилигандными электронными переходами (рис. 3).
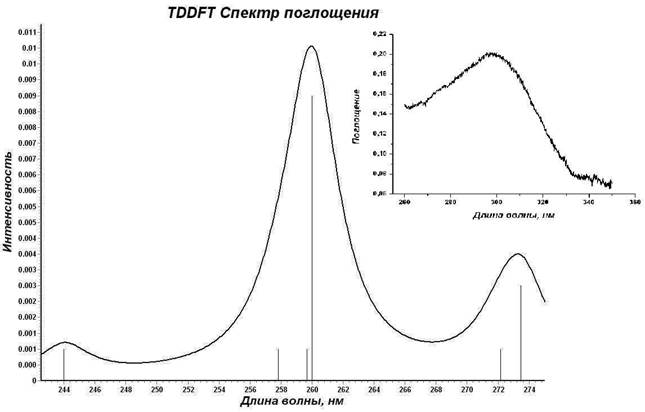
Рис. 3. Смоделированный спектр поглощения комплекса La(NO3)3(ГМФА)3 (на вкладке – экспериментальный спектр поглощения [4])
По данным расчетов, электронные синглет-синглетные переходы молекулярной системы из возбужденного в основное состояние обусловлены переходами валентных электронов с вакантных молекулярных уровней, соответствующих несвязывающей π*-МО π–системы групп NO3, на молекулярные уровни НЭП атомов азота нейтральных лигандов ГМФА. Таким образом, полоса флуоресценции с максимальной интенсивностью (λфлуор = 440 нм) экспериментального спектра люминесценции комплекса I также обусловлена внутрилигандными электронными переходами (рис. 4).
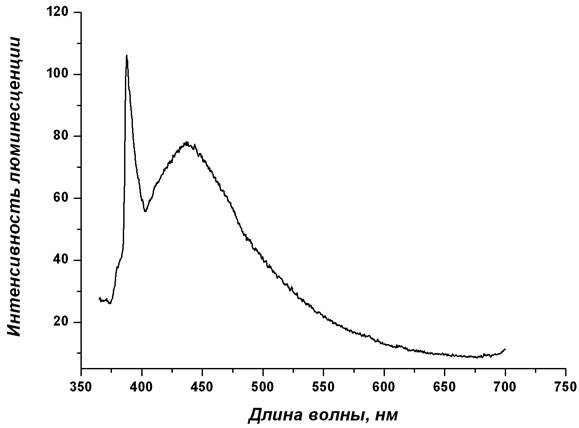
Рис. 4. Экспериментальный спектр люминесценции La(NO3)3(ГМФА)3 [4]
Квантово-химические расчеты параметров электронных триплет-синглетных переходов показали, что электронные переходы соединения I из возбужденного триплетного в основное синглетное состояние более чем на 80% связано с электронными переходами с вакантных молекулярных уровней, соответствующих несвязывающей π*-МО π–системы групп NO3, на молекулярные уровни, соответствующие НЭП атомов кислорода групп NO3.
Выводы
Методами функционала плотности DFT и TDDFT/PBE0/ECP-MWB в вакуумном приближении смоделированы структура и спектральные свойства комплекса лантана (III) La(NO3)3(ГМФА)3. Выполнены квантово-химические расчеты геометрического и электронного строения данного хелатного комплексного соединения в основном и возбужденных состояниях. Показано, что данная молекулярная система является полярной, ее дипольный момент равен 9.2 Д. По результатам моделирования дана интерпретация особенностей экспериментальных электронных спектров поглощения и люминесценции.
Выявлен эффект дислокации в кристалле на электронное строение соединения. С целью определения механизма ТЛ в хелатных соединениях РЗЭ состава Ln(NO3)3ГМФА3 (Ln = Eu, Tb) проведено квантово-химическое моделирование кластеров (La(NO3)3ГМФА3)n (n = 2–4). Теоретическое изучение показало, что при смещении слоев молекул относительно друг друга в кластере происходит значительное изменение термодинамических параметров молекулярной системы, увеличение ее полной энергии, существенная поляризация системы (дипольный момент тетрамера возрастает от 0.0 до 7.32 Д).
По результатам моделирования сделаны выводы о механизме люминесценции, обсуждена возможность управления люминесценцией в наноструктурированных материалах. Дана интерпретация особенностей экспериментальных электронных спектров поглощения и спектров люминесценции.
Работа проводилась при частичной финансовой поддержке Министерства образования и науки Российской Федерации в рамках государственного задания Дальневосточного федерального университета № 3.2261.2011.
Рецензенты:
Игнатьева Лидия Николаевна, д.х.н., заведующая лабораторией фторидных материалов, ФГБУН «Институт химии ДВО РАН», г. Владивосток.
Кавун Валерий Яковлевич, д.х.н., заведующий лабораторией химической
радиоспектроскопии, ФГБУН «Институт химии ДВО РАН», г. Владивосток.