


Scientific journal
Modern problems of science and education
ISSN 2070-7428
"Перечень" ВАК
ИФ РИНЦ = 0,936
DEVELOPMENT AND VALIDATION OF DESMETHYLSIBUTRAMINE QUANTITATIVE DETERMINATION METHOD BY GLC-FID

Проблема злоупотребления психоактивными веществами в России из года в год становится все более актуальной. В последнее время участились случаи обнаружения сибутрамина и его активных метаболитов (М1 и М2) в биологически активных добавках для похудения (БАД). Федеральная Служба РФ по контролю за оборотом наркотиков в Методических рекомендациях по криминалистическому исследованию сибутрамина от 2006 г. сообщала, что, с учетом дофаминовой теории формирования наркологических заболеваний, сибутрамин может вызывать привыкание и зависимость, а клинические описания его действия позволяют предположить использование его потребителями психоактивных веществ в качестве психостимулятора [4]. С 2008 года сибутрамин, а также его структурные аналоги, обладающие схожим психоактивным действием, включены в список сильнодействующих веществ для целей статьи 234 и других статей Уголовного кодекса Российской Федерации [3].
Сибутрамин метаболизируется изоферментом CYP3А4 до деметилированных метаболитов M1 и М2 (моно- и дидесметилсибутрамин), которые обуславливают его терапевтический эффект и являются структурными аналогами сибутрамина [6, 7, 8]. Установлено, что десметилсибутрамин (ДМС) и дидесметилсибутрамин (ДДМС) примерно в 100 раз активнее исходного соединения [5, 8].
Согласно действующему законодательству РФ, сильнодействующие вещества и их аналоги запрещены к использованию в составе БАД [2]. Ввиду сложившейся ситуации вопрос об усовершенствовании процедуры обнаружения посторонних токсикологически важных веществ, не заявленных в составе БАД, стоит достаточно остро. Однако на сегодняшний день в литературных источниках отсутствует какая-либо информация по процедуре контроля БАД на наличие сильнодействующих веществ, в то время как в практике судебно-химических исследований имеются факты обнаружения данных веществ в биологически активных добавках к пище.
Цель исследования
Разработка методики количественного определения активного метаболита сибутрамина, ДМС, методом газо-жидкостной хроматографии (ГЖХ).
Материал и методы исследования
Субстанцию ДМС получали из содержимого капсул «Жуйдэмэн» (500 мг, № 60), содержащих данное вещество, при комнатной температуре следующим образом: содержимое капсул высыпали в чистую стеклянную склянку, залили спиртом этиловым 95% (1:10), встряхивали в течение 15 минут, затем надосадочную жидкость сливали в чистую стеклянную склянку. Оставшийся осадок вновь заливали спиртом этиловым 95% и операции повторяли. Надосадочные жидкости объединяли и выпаривали без нагревания до сухого остатка. Получившийся сухой остаток брался из расчета, что его состав соответствует экстракту содержимого одной капсулы. После перекристаллизации экстракт был проанализирован на газовом хроматографе Agilent 7890A с масс-спектрометром Agilent 5975C, в результате чего была идентифицирована и подтверждена его химическая структура: масс-спектр пика со временем удерживания 13,18 мин соответствовал библиотечному масс-спектру ДМС (рис. 1).
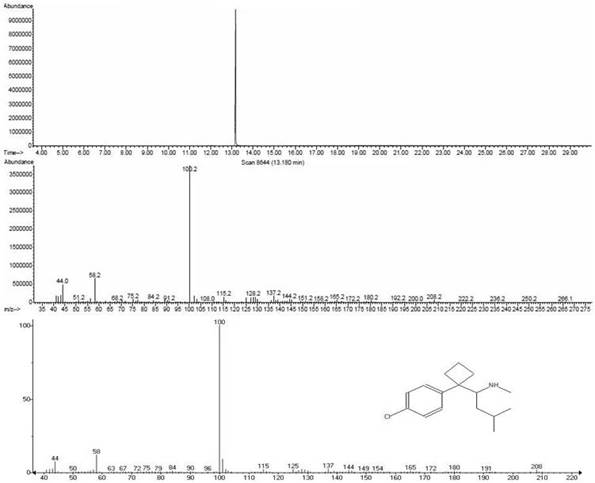
Рисунок 1. Хроматограмма перекристаллизованного экстракта (верхняя часть рисунка), масс-спектр пика с временем удерживания 13,18 мин (средняя часть рисунка) и библиотечный масс-спектр десметилсибутрамина (нижняя часть рисунка).
Готовили растворы ДМС в этиловом 96 % спирте в концентрациях 1 мг/мл, 500 мкг/мл, 200 мкг/мл, 100 мкг/мл, 50 мкг/мл и 10 мкг/мл. В качестве внутреннего стандарта использовали раствор метилстеарата в 96 % этиловом спирте в концентрации 1 мг/мл.
Исследование проводили на газовом хроматографе Хроматэк-Кристалл 5000 с пламенно-ионизационным детектором в следующих условиях: колонка HP-5MS, скорость потока газа-носителя (азот) 2,3 мл/мин, температура термостата колонки начальная 170 °С, конечная – 220 °С, температура детектора 250 °С, температура испарителя 230 °С, ввод пробы с делением потока 1/3, объем вводимой пробы 1 мкл, время хроматографирования 18 мин.
Результаты исследования и их обсуждение
Количественное определение ДМС осуществляли методом ГЖХ с расчетом концентрации по методу внутреннего стандарта, в качестве которого был выбран метилстеарат, преимуществами использования которого являются его хроматографические свойства, близкие к определяемому веществу, стабильность полученных результатов, и доступность для закупки на территории РФ.
Время удерживания ДМС и метилстеарата в заданных условиях хроматографирования составило 7,07 мин и 10,93 мин соответственно (рис. 2).
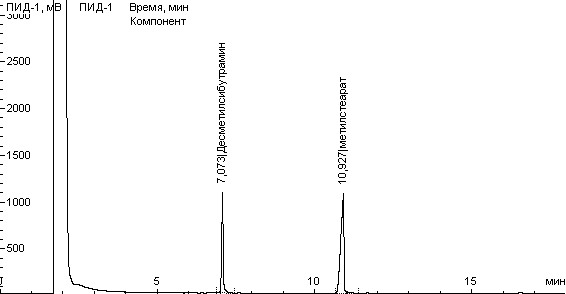
Рисунок 2. Хроматограмма раствора ДМС с внутренним стандартом (метилстеарат) в концентрации 1000 мкг/мл.
Валидацию разработанной методики осуществляли по показателям: линейность, правильность, прецизионность (на уровне intra-day и inter-day), аналитическая область [1].
Линейность
Для определения линейности проводили анализ 6 калибровочных спиртовых растворов ДМС с концентрациями от 10 мкг/мл до 1000 мкг/мл в присутствии внутреннего стандарта (1000 мкг/мл) (табл. 1).
Таблица 1
Значения концентраций калибровочных растворов и отношений площадей пиков ДМС к площадям пиков метилстеарата
|
СДМС, мкг/мл |
СМС, мкг/мл |
SДМС |
SМС |
SДМС/ SМС |
|||
|
Si |
Sср |
Si |
Sср |
Si |
Sср |
||
|
10 |
1000 |
49,919 |
46,512 |
8319,833 |
7752,000 |
0,0060 |
0,0060 |
|
45,663 |
7610,500 |
0,0060 |
|||||
|
43,954 |
7325,667 |
0,0060 |
|||||
|
50 |
1000 |
193,912 |
200,776 |
6002,845 |
6078,020 |
0,0323 |
0,0330 |
|
211,794 |
6310,256 |
0,0336 |
|||||
|
196,621 |
5920,958 |
0,0332 |
|||||
|
100 |
1000 |
303,652 |
405,149 |
5855,909 |
7692,778 |
0,0519 |
0,0526 |
|
489,749 |
9227,440 |
0,0531 |
|||||
|
422,047 |
7994,986 |
0,0528 |
|||||
|
200 |
1000 |
677,061 |
678,460 |
5732,946 |
5754,650 |
0,1181 |
0,1179 |
|
681,875 |
5808,131 |
0,1174 |
|||||
|
676,443 |
5722,872 |
0,1182 |
|||||
|
500 |
1000 |
1550,308 |
1870,566 |
5834,157 |
6914,314 |
0,2660 |
0,2703 |
|
2129,204 |
7808,231 |
0,2730 |
|||||
|
1932,187 |
7100,553 |
0,2720 |
|||||
|
1000 |
1000 |
3814,719 |
3619,324 |
6320,993 |
5998,512 |
0,6035 |
0,6034 |
|
3286,095 |
5446,867 |
0,6033 |
|||||
|
3757,157 |
6227,676 |
0,6033 |
|||||
*СДМС – концентрация десметилсибутрамина в растворе, СМС – концентрация метилстеарата в растворе,
SДМС – площадь пика ДМС, SМС – площадь пика метилстеарата.
По полученным значениям строили график линейной зависимости. Были рассчитаны коэффициенты регрессионной прямой у = 0,0006х методом наименьших квадратов, где у – среднее значение отношения площади пика ДМС к площади пика метилстеарата, рассчитанное по трем хроматограммам, х – концентрация ДМС, мкг/мл (Сфакт). (рис. 3).
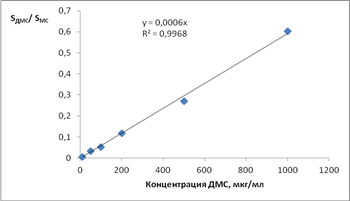
Рисунок 3. Калибровочный график зависимости отношения площади пика ДМС к площади пика метилстеарата от концентрации ДМС в растворе
Квадрат линейного коэффициента корреляции (R2) характеризует степень соответствия между регрессионной моделью и исходными данными. В данном случае 99,68% изменений зависимой переменной описывается регрессионным уравнением. Коэффициент корреляции R = 0,9984, что свидетельствует о наличии прямой линейной зависимости между площадью пика ДМС и его концентрацией в растворе.
Правильность и прецизионность
Для оценки правильности и прецизионности методики проводили анализ 3 калибровочных спиртовых растворов ДМС с концентрациями 10 мкг/мл, 200 мкг/мл, и 1000 мкг/мл в течение первого дня (intra-day) и второго дня (inter-day). Каждый раствор хроматографировали в трех повторностях. Для полученных значений концентрации ДМС рассчитывали величину стандартного отклонения (SD), относительного стандартного отклонения (RSD, %) и отклонение от заданной величины (ε, %). Данные представлены в таблицах 2 и 3.
Таблица 2
Оценка правильности и прецизионности (intra-day)
|
Сфакт, мкг/мл |
Сизм, мкг/мл |
Сср (n=3) |
SD (n=3) |
RSD, % (n=3) |
ε, % |
|
10 |
10,03 |
10,04 |
0,01 |
0,12 |
0,40 |
|
10,03 |
|||||
|
10,05 |
|||||
|
200 |
196,85 |
197,08 |
0,24 |
0,12 |
1,46 |
|
197,33 |
|||||
|
197,07 |
|||||
|
1000 |
1005,83 |
1005,61 |
0,19 |
0,02 |
0,56 |
|
1005,50 |
|||||
|
1005,50 |
*Сфакт – фактическая концентрация ДМС в растворе, Сизм – концентрация ДМС в растворе, рассчитанная по методике.
Таблица 3
Оценка правильности и прецизионности (inter-day)
|
Сфакт, мкг/мл |
Сизм, мкг/мл |
Сср (n=3) |
SD (n=3) |
RSD, % (n=3) |
ε, % |
|
10 |
9,87 |
9,91 |
0,05 |
0,52 |
0,90 |
|
9,90 |
|||||
|
9,97 |
|||||
|
200 |
195,33 |
195,61 |
0,56 |
0,29 |
2,19 |
|
195,25 |
|||||
|
196,25 |
|||||
|
1000 |
1007,83 |
1007,81 |
0,28 |
0,03 |
0,78 |
|
1007,52 |
|||||
|
1008,08 |
*Сфакт – фактическая концентрация ДМС в растворе, Сизм – концентрация ДМС в растворе, рассчитанная по методике.
Полученные значения RSD и ε свидетельствуют о достаточной степени соответствия между истинным значением определяемого вещества и его значением, рассчитанным по данной методике.
Аналитическая область методики
Аналитическая область методики на основании результатов оценки линейности, правильности и прецизионности составила 10 – 1000 мкг/мл.
Заключение
Разработана методика количественного определения десметилсибутрамина методом газовой хроматографии с пламенно-ионизационной детекцией, обладающая необходимой линейностью, правильностью и прецизионностью, что позволяет говорить о хороших валидационных характеристиках данной методики.
Рецензенты:
Вихарева Е.В., д.фарм.н., доцент, заведующий кафедрой аналитической химии ГБОУ ВПО ПГФА Минздрава России, г. Пермь;
Ярыгина Т.И., д.фарм.н., профессор кафедры фармацевтической химии факультета очного обучения ГБОУ ВПО ПГФА Минздрава России, г. Пермь.
Библиографическая ссылка
Стерн К.И., Малкова Т.Л. РАЗРАБОТКА И ВАЛИДАЦИЯ МЕТОДИКИ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДЕСМЕТИЛСИБУТРАМИНА МЕТОДОМ ГЖХ С ПЛАМЕННО-ИОНИЗАЦИОННЫМ ДЕТЕКТИРОВАНИЕМ // Современные проблемы науки и образования. 2014. № 6. ;URL: https://science-education.ru/en/article/view?id=15483 (дата обращения: 25.07.2026).