



Введение
Диарилпроизводные пиримидиновых гетероциклов являются интересным классом органических соединений, обладающих биологической активностью различных типов. Среди них известно множество соединений, способных ингибировать репродукцию оболочечных вирусов, таких как вирус гепатита C [7] и в особенности вирус иммунодефицита человека (ВИЧ-1) [4; 8]. Одними из наиболее интересных объектов для введения ароматических остатков является урацил и его производные благодаря разнообразию синтетических методологий, позволяющих проводить их функционализацию.
Ранее нами был описан синтез производных урацила, содержащих ароматические фрагменты, связанные короткими мостиковыми фрагментами с гетероциклическим ядром в положениях 1 и 6 [1] (рис. 1).
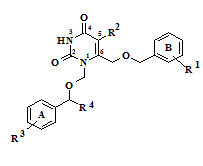
Рисунок 1 – Общая структура 1,6-бис[(бензилокси)метил]производных урацила.
Целью настоящего исследования является изучение активности в отношении обратной транскриптазы ВИЧ-1 и ряда ее мутантных форм, содержащих аминокислотные замены в участке связывания ненуклеозидных ингибиторов, а также анализ закономерностей «структура-активность» в ряду 1,6-бис[(бензилокси)метил]производных урацила.
Материалы и методы
В работе использовались следующие материалы: [α-32P]dATP (5000 Ки/моль) фирмы «Изотоп» (Россия), 2`-дезоксирибонуклеозид–5’–трифосфаты (Promega, США), целлюлозные фильтры Whatman 3MM (Whatman, Великобритания). Все остальные реактивы максимальной чистоты были приобретены у компаний Sigma-Aldrich или Fluka. Активированная ДНК была получена из ДНК спермы лосося (Pharmacia Biotech, США) обработкой ДНКазой поджелудочной железы быка (Fermentas, Литва), как описано в работе [2].
Определение активности ОТ ВИЧ-1 в системе активированной ДНК. Стандартная реакционная смесь (20 мкл) содержала 150 мкг/мл активированной ДНК, 0,05 мкг ОТ ВИЧ-1, 3 мкМ ATP, по 30 мкМ остальных нуклеозид-5`-трифосфатов, 0,02 МБк [α-32P]dATP в буфере для измерения активности ОТ ВИЧ-1 (50 мМ Tрис•HCl, pH 8,1, 10 мМ MgCl2 и 200 мМ KCl). В экспериментах по исследованию ингибиторных свойств соединения вносили в реакционную смесь в виде растворов в диметилсульфоксиде до конечной концентрации последнего, равной 10%, при этом к контрольным реакциям прибавляли аналогичный объем чистого ДМСО. Реакцию инициировали прибавлением обратной транскриптазы и инкубировали в течение 20 мин при 37 °C, затем наносили пробы на фильтры (1x1 см) Whatman 3MM, пропитанные 1 мкл 0,5 M раствора ЭДТА. Фильтры отмывали от не включившегося в ДНК меченого нуклеотида 5 х 25 мл 10%-ной трихлоруксусной кислоты в течение 5 мин каждый, 25 мл этилового спирта и сушили на воздухе. Сорбированную на фильтрах радиоактивность измеряли по методу Черенкова в счетчике Intertechnique Liquid Scintillation Counter SL-4000. Расчет констант ингибирования проводили по методу Диксона для неконкурентного типа ингибирования [6].
Результаты и их обсуждение
Анализ результатов исследования биологической активности синтезированных соединений – 1,6-бис[(бензилокси)метил]производных урацила 1-14 и их N1-1-(3-метилбутокси)метильных аналогов 15-16 позволил выявить закономерности «структура-активность» в данном ряду веществ. Наиболее активным в отношении обратной транскриптазы (ОТ) ВИЧ-1 дикого типа оказался 6-[(бензилокси)метил]-1-[(3,5-дихлорбензилокси)метил]урацил (10), ингибировавший на 50% активность фермента в концентрации 0,62 μM и превосходивший «родоначальное» соединение ряда (1) по активности более, чем в два раза. Введение метильных заместителей в положения 3 и 5 цикла A (11), а также цианогруппы в положение 4 данного фрагмента (12) приводило к снижению активности, в то время как введение заместителей в положение 4 ароматического фрагмента B практически не оказывало значимого влияния на активность (2-3). Введение атомов галогенов в положение 5 гетероцикла (5-9) во всех случаях снижало активность по отношению к 5-незамещенным аналогам. Разветвленные [(1-фенилэтокси)метил]производные (13-14) не проявили активности в отношении ОТ, а алифатические аналоги (15-16) показали на порядок более низкую активность, чем ароматические производные. Их константы ингибирования составили 33,4 и 50,4 μМ, соответственно (таблица 1).
Таблица 1 – Активность соединений 1-16 в отношении ОТ ВИЧ-1 дикого типа
1-14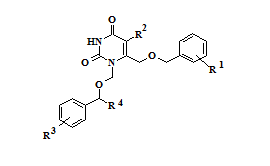
 15-16
15-16
|
Соед. |
R1 |
R2 |
R3 |
R4 |
Ki (µМ) |
|
1 |
H |
H |
H |
H |
1,4 |
|
2 |
4-Cl |
H |
H |
H |
1,66 |
|
3 |
4-CH3 |
H |
H |
H |
2,13 |
|
4 |
3,5-Cl2 |
H |
H |
H |
ND* |
|
5 |
H |
Cl |
H |
H |
8,0 |
|
6 |
H |
Br |
H |
H |
15 |
|
7 |
4-Cl |
Br |
H |
H |
16,9 |
|
8 |
4-CH3 |
Br |
H |
H |
12,9 |
|
9 |
H |
I |
H |
H |
8,43 |
|
10 |
H |
H |
3,5-Cl2 |
H |
0,62 |
|
11 |
H |
H |
3,5-(CH3)2 |
H |
1,67 |
|
12 |
H |
H |
4-CN |
H |
50 |
|
13 |
H |
H |
H |
CH3 |
>100 |
|
14 |
4-Cl |
H |
H |
CH3 |
>100 |
|
15 |
H |
- |
- |
- |
33,4 |
|
16 |
4-Cl |
- |
- |
- |
50,4 |
|
Невирапин |
7,2 |
||||
* активность не определена
Ki (константа ингибирования) – концентрация ненуклеозидного ингибитора обратной транскриптазы, обеспечивающая подавление активности фермента на 50%.
Анализ ингибиторной активности соединений 1-3 и 10-11 в отношении ряда мутантных форм ОТ показал, что исследуемые вещества оказываются на 1-2-порядка менее активными в отношении всех исследуемых типов фермента в сравнении с ОТ дикого типа. Исследованные вещества превосходили по активности используемый в клинической практике антиретровирусный препарат невирапин (Viramune®) в отношении одиночных мутантов и двойного мутанта K103N/Y181C, однако значительно уступали эфавиренцу по действию на те же формы ОТ. Наиболее благоприятный профиль резистентности по совокупности данных продемонстрировал 6-[(бензилокси)метил]-1-[(3,5-дихлорбензилокси)метил]урацил (10). Интересной особенностью данного соединения явилась высокая активность в отношении ОТ с мутацией G190A, превышающая таковую для невирапина более чем в 217 раз. Следует также отметить, что наиболее выраженную активность в отношении одного из важнейших клинических одиночных мутантов вируса Y181C показало соединение 2, превышая эффективность невирапина более чем в 440 раз. Лидером по активности в отношении двойного мутантного изолята вируса K103N/Y181C стал 1-[(бензилокси)метил]-6-[(4-хлорбензилокси)метил]урацил (2) (таблица 2).
Таблица 2 – Активность соединений 1-3 и 10-11 в отношении мутантных вариантов ОТ ВИЧ-1
|
ОТ ВИЧ-1 |
1 |
2 |
3 |
10 |
11 |
Невирапин |
Эфавиренц |
|
Ki, μM |
|||||||
|
дикий тип |
1,4 |
1,66 |
2,13 |
0,62 |
1,67 |
7,2 |
0,01 |
|
L100I |
189 |
69 |
77 |
51 |
125 |
273 |
0,08 |
|
K103N |
56 |
51 |
67 |
38 |
124 |
>2000 |
0,58 |
|
V106A |
123 |
105 |
67 |
41 |
131 |
>2000 |
0,05 |
|
Y181C |
60,6 |
4,5 |
10 |
26 |
109 |
>2000 |
0,03 |
|
G190A |
>140 |
>166 |
>213 |
9,2 |
41 |
>2000 |
0,06 |
|
K103N/Y181C |
>140 |
61 |
67 |
>62 |
>167 |
>2000 |
0,14 |
Ki (константа ингибирования) – концентрация ненуклеозидного ингибитора обратной транскриптазы, обеспечивающая подавление активности фермента на 50%.
Для рационализации полученных данных был проведен молекулярный докинг некоторых из полученных соединений в структуру комплексов ОТ с известными ННИОТ, депонированными в Банке данных белков (PDB-коды 3C6T и 4H4M) [3; 5]. Наибольшее отрицательное значение свободной энергии связывания лидера в ряду производных 1,6-бис[(бензилокси)метил]урацила (соединение 10) было достигнуто в случае комплекса 3С6T (исходный лиганд - 2-[3-хлор-5-(3-хлор-5-цианофенокси)фенокси]-N-(2-хлор-4-сульфамоил-фенил)ацетамид). Обнаружено близкое соответствие расположения 3,5-дихлорбензильного фрагмента соединения 10 и цикла А исходного диарилэфирного ННИОТ. Согласно результатам докинга, данный фрагмент образует π-стэкинг-взаимодействия с остатками аминокислот Y181, Y188 и W229. Благоприятный эффект от введения мета-заместителей на ингибирование активности фермента может быть связан с продвижением цикла А глубже в область гидрофобного кармана, формируемую ароматическими аминокислотными остатками, а также образованием дополнительных Ван-дер-Ваальсовых взаимодействий с остатками L234 и F227 [5; 9]. Отрицательное влияние атомов галогенов в положении 5 на активность может объясняться электростатическим отталкиванием между ними и атомом кислорода мостиковой цепочки в положении 6, приводящем к изменению оптимальной конформации последней. Атом кислорода амидной группы остатка урацила способен к формированию водородной связи с NH-фрагментом остатка K103, при этом расстояние между центрами атомов О и N оценивается в 3,2 Å, что несколько больше (2,8 Å) расстояния между атомом кислорода карбонильной группой оксиацетамидного фрагмента модельного ингибитора и атомом азота в том же аминокислотном остатке фермента, однако не превышает дистанцию Ван-дер-Ваальса. Согласно результатам докинга, возможно также формирование дополнительной водородной связи между атомом азота в амидном фрагменте урацила (N3) и амидным функцией остатка K101 (рисунок 2).
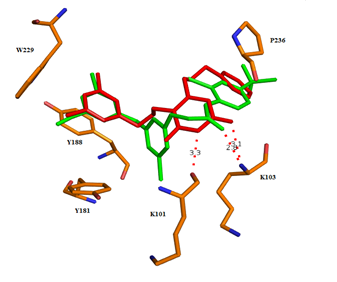
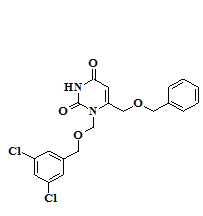
Рисунок 2 – Возможная конформация
6-[(бензилокси)метил]-1-[(3,5-дихлорбензилокси)метил]урацила (10, выделен красным) в наложении с диарилэфирным ННИОТ (PDB-код 3C6T, выделен зеленым) в аллостерическом сайте ОТ дикого типа ВИЧ-1
Выводы
Среди производных 1,6-бис[(бензилокси)метил]урацила были обнаружены высокоактивные ингибиторы обратной транскриптазы ВИЧ-1, проявляющие активность на субмикромолярном уровне. Полученные соединения являются перспективными для дальнейшего изучения и внедрения в практику.
Настоящая работа выполнена при поддержке гранта Российского фонда фундаментальных исследований (№ 13-04-01391).
Рецензенты:
Ганичева Л.М., д.фарм.н., доцент кафедры управления и экономики фармации, медицинского и фармацевтического товароведения Волгоградского государственного медицинского университета, г. Волгоград.
Симонян А.В., д.фарм.н., профессор кафедры фармацевтической технологии и биотехнологии Волгоградского государственного медицинского университета, г. Волгоград.